模拟计算服务
专业的计算模拟服务,涵盖第一性原理、量子化学、分子动力学、生物模拟和机器学习
第一性原理计算
基于量子力学第一性原理的材料性质计算,提供从电子结构到宏观性能的全面分析
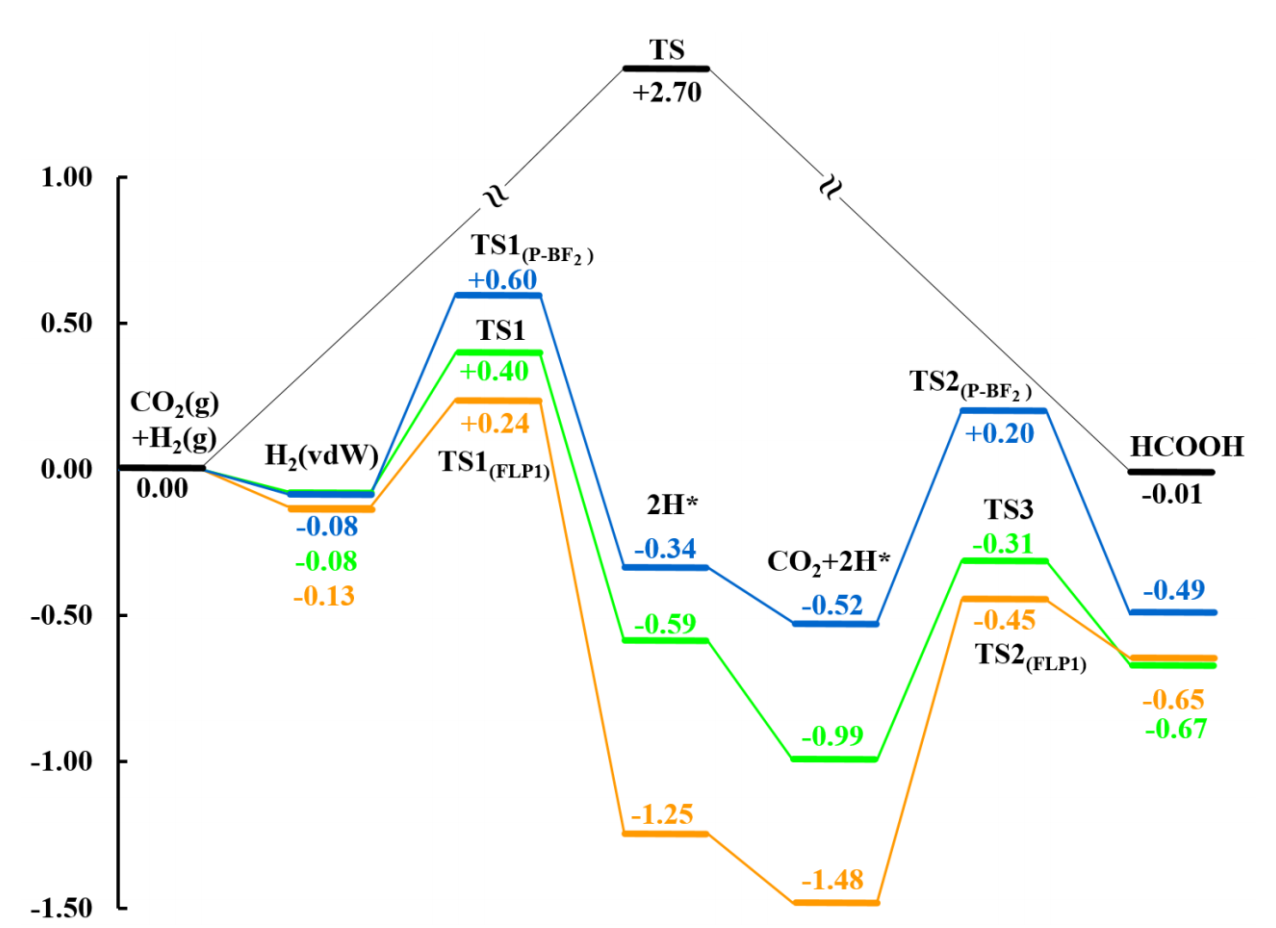
自由能台阶图
能够体现催化反应进行的难易程度。台阶图顾名思义,因它与台阶类似的形状而得名,是在电催化计算中最常见的一种表现形式。
迁移能垒
能垒越低,离子的迁移能力越强。在锂离子、钠离子等电池体系中,离子在电极材料中的迁移扩散难易程度是体现电池性能的重要因素。
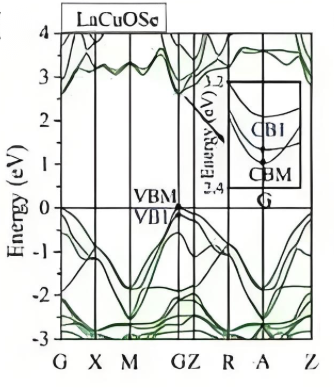
能带
体现结构的电子性质的常用方法之一。能带结构是指材料中所有电子能级的分布情况,包括价带和导带。
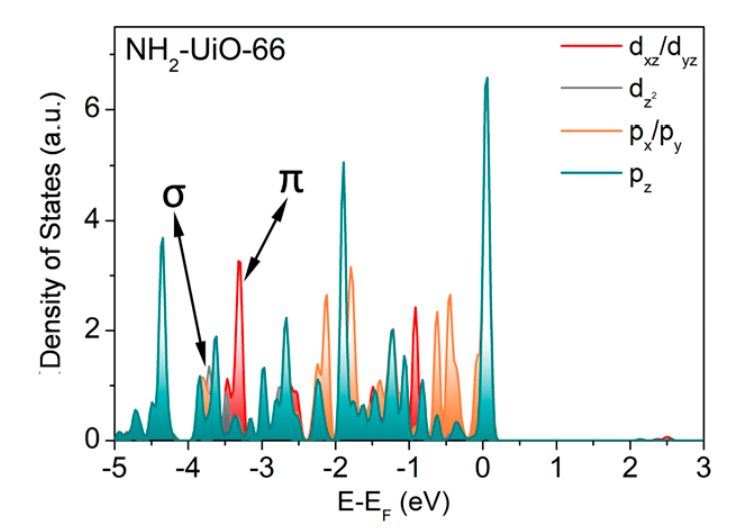
态密度
态密度可以反应结构的成键信息等。态密度表示单位能量范围内的电子数目,从态密度图中可以得到结构的成键信息。
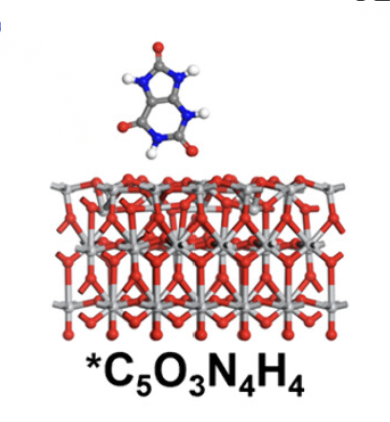
吸附能
体现分子在表面的吸附能力的强弱。吸附能是指分子或原子在材料表面吸附时释放或吸收的能量。
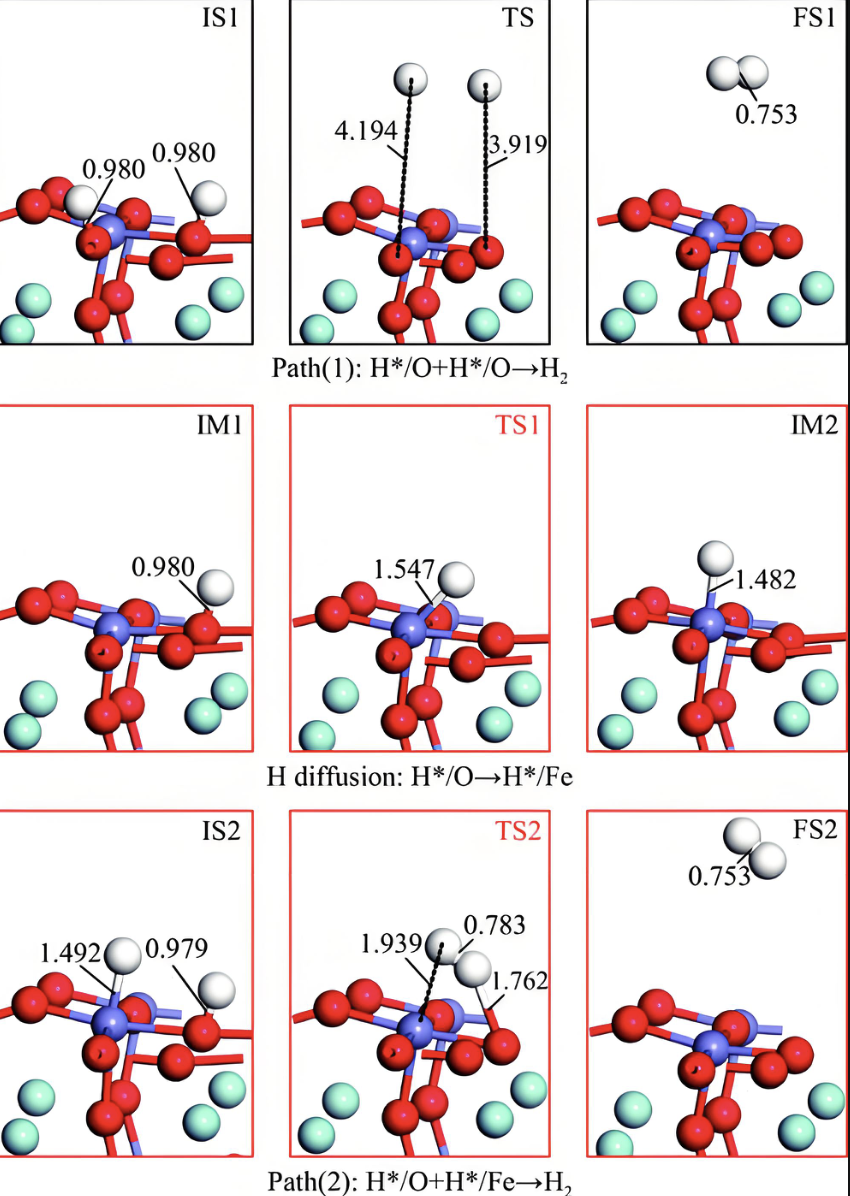
过渡态
能垒的高低体现了反应的难易程度。过渡态是指在反应物生成最终产物的过程中,可能会存在的一个能量最高但不稳定的中间态。
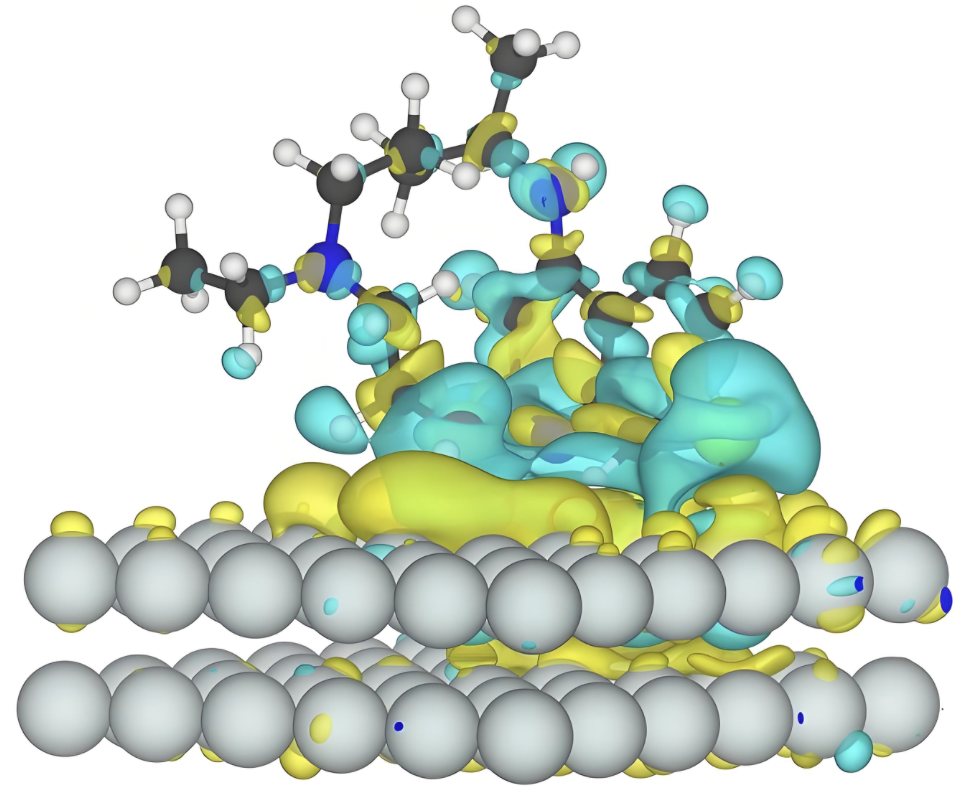
差分电荷密度
定性分析结构之间的电荷转移情况。差分电荷密度是研究电子结构的重要手段之一,可以直观得到催化剂和吸附中间体之间的电子相互作用。
功函数
其大小标志着束缚电子的强弱,功函数越小,电子越容易逸出材料表面。是表征材料电子发射能力的重要参数。
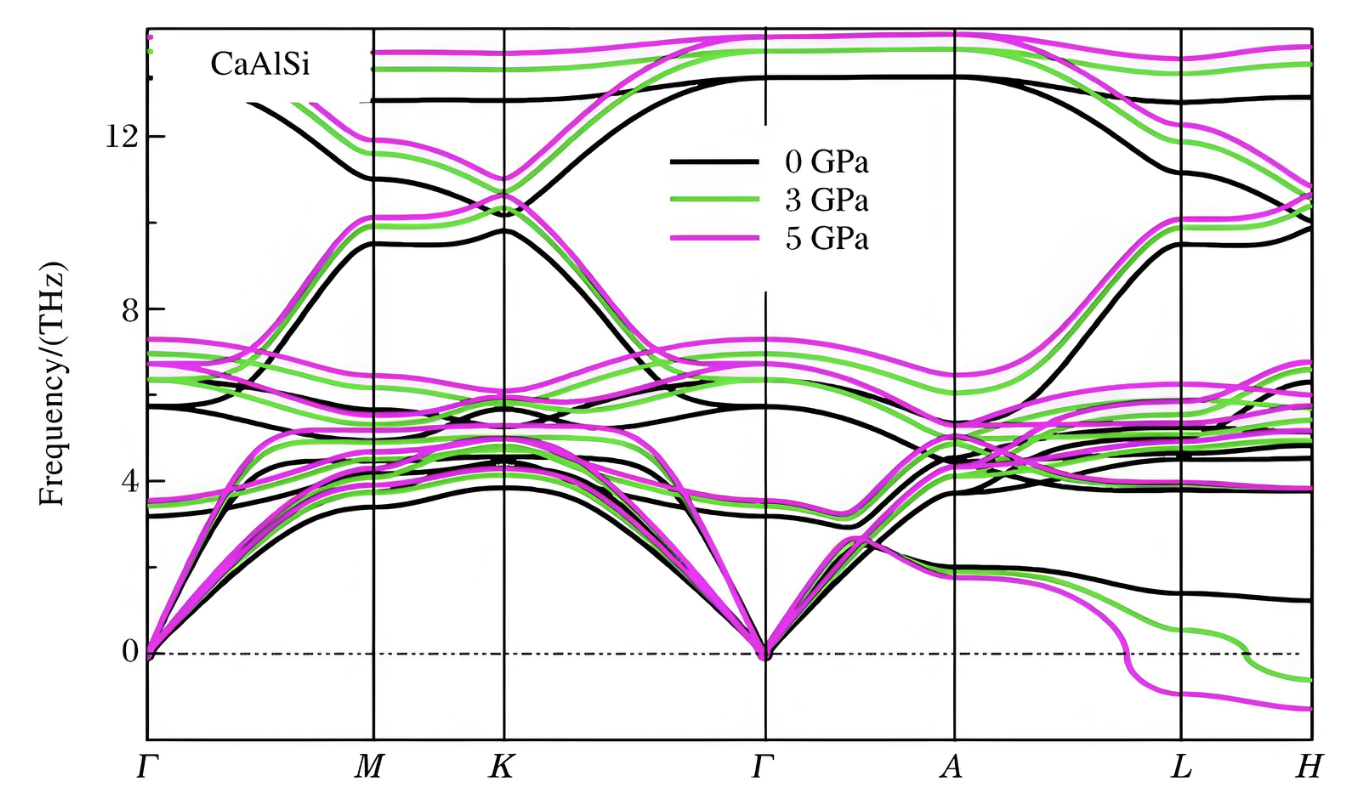
声子谱
声子谱可以分析结构的稳定性。声子用来描述晶格的简谐振动,声子谱是研究材料热力学性质的重要切入点。
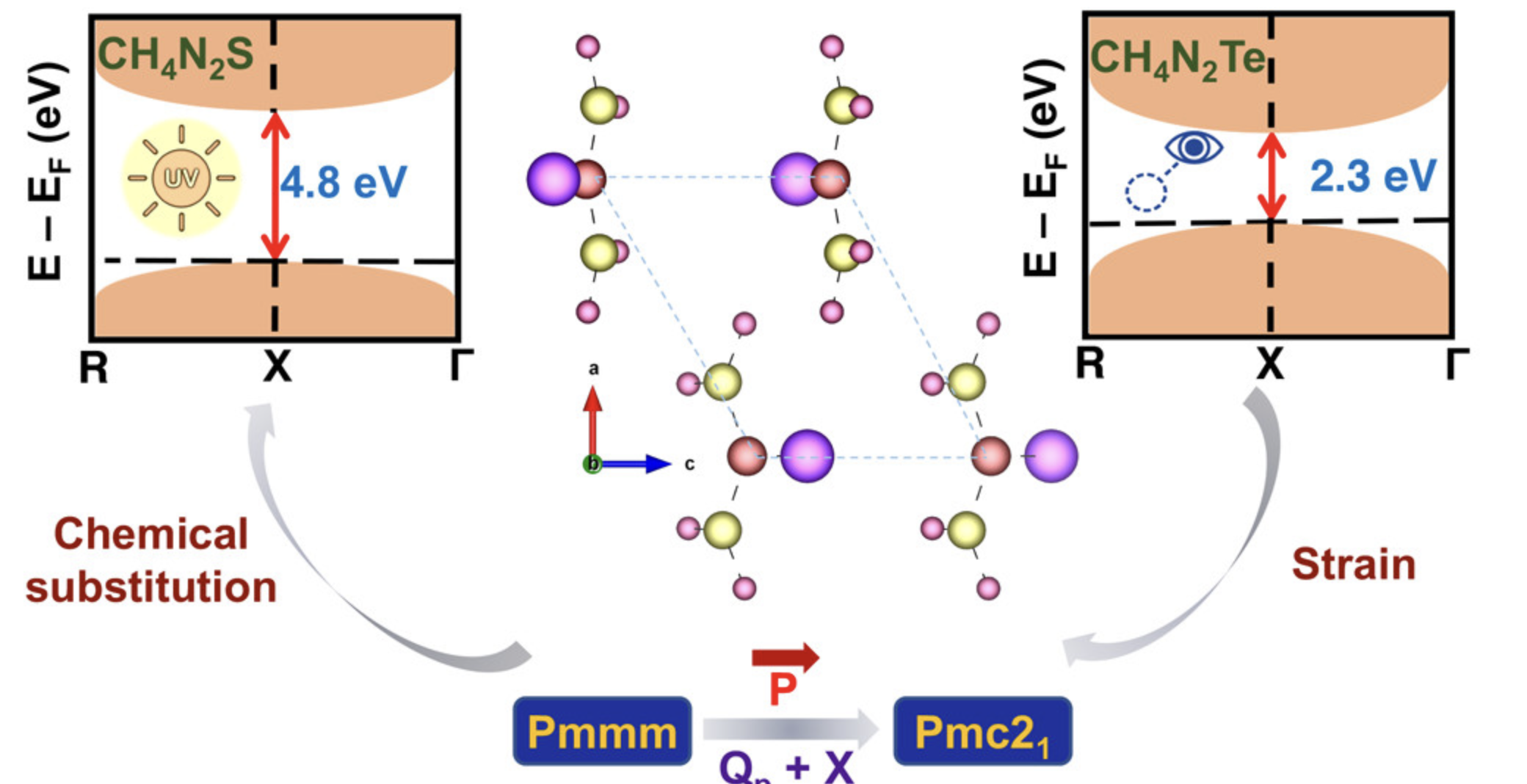
光学性质
揭示光与物质相互作用的本质。材料的光学性质决定了不同能量的光子在材料中的透射、折射、散射和吸收等性质。
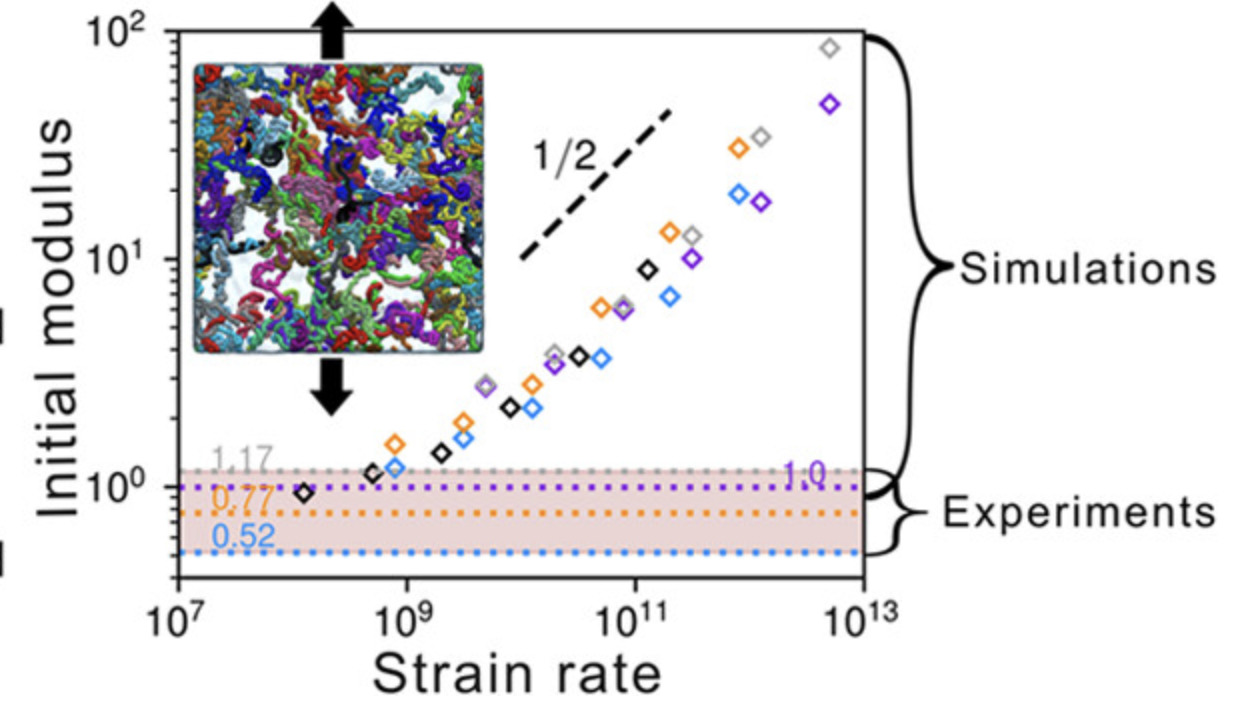
弹性性质
体现结构在外力下的稳定性。弹性性质是指材料在受到外力作用后发生形变,并在去除外力后能够恢复原状的性质。
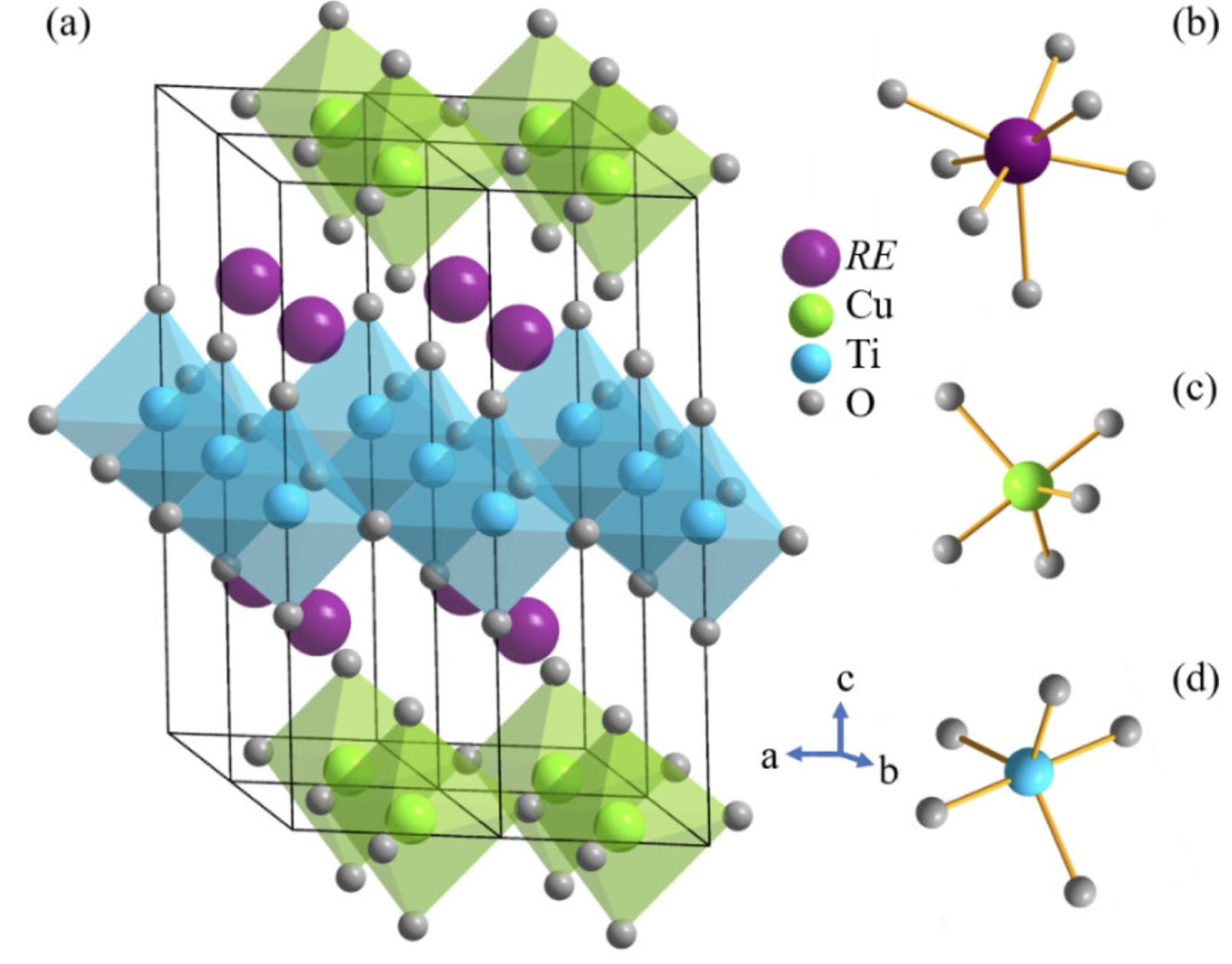
磁性质
体现结构的磁各向异性、磁导率等。磁性质是指物质在磁场中表现出的一系列性质,包括磁化、磁导率、磁滞、磁各向异性等。
空位形成能
形成能越小,空位结构越容易形成。通过在结构中去除一部分原子,形成空位结构,以改变原来的结构中的相互作用。
火山图
理解催化活性与结构性质之间的关系。火山图是一种用来描述催化反应速率与反应物浓度之间关系的图表。
COOP/COHP
分析结构中化学键的强度。COOP(晶体轨道重叠布局)和COHP(晶体轨道哈密顿布居)是分析化学键和晶体结构电子特性的重要工具。
电子局域密度函数
电子在空间中的分布和局域化程度。电子局域密度函数量化了电子对的局域化程度,可以区分共价键、离子键和金属键等不同的电子环境。
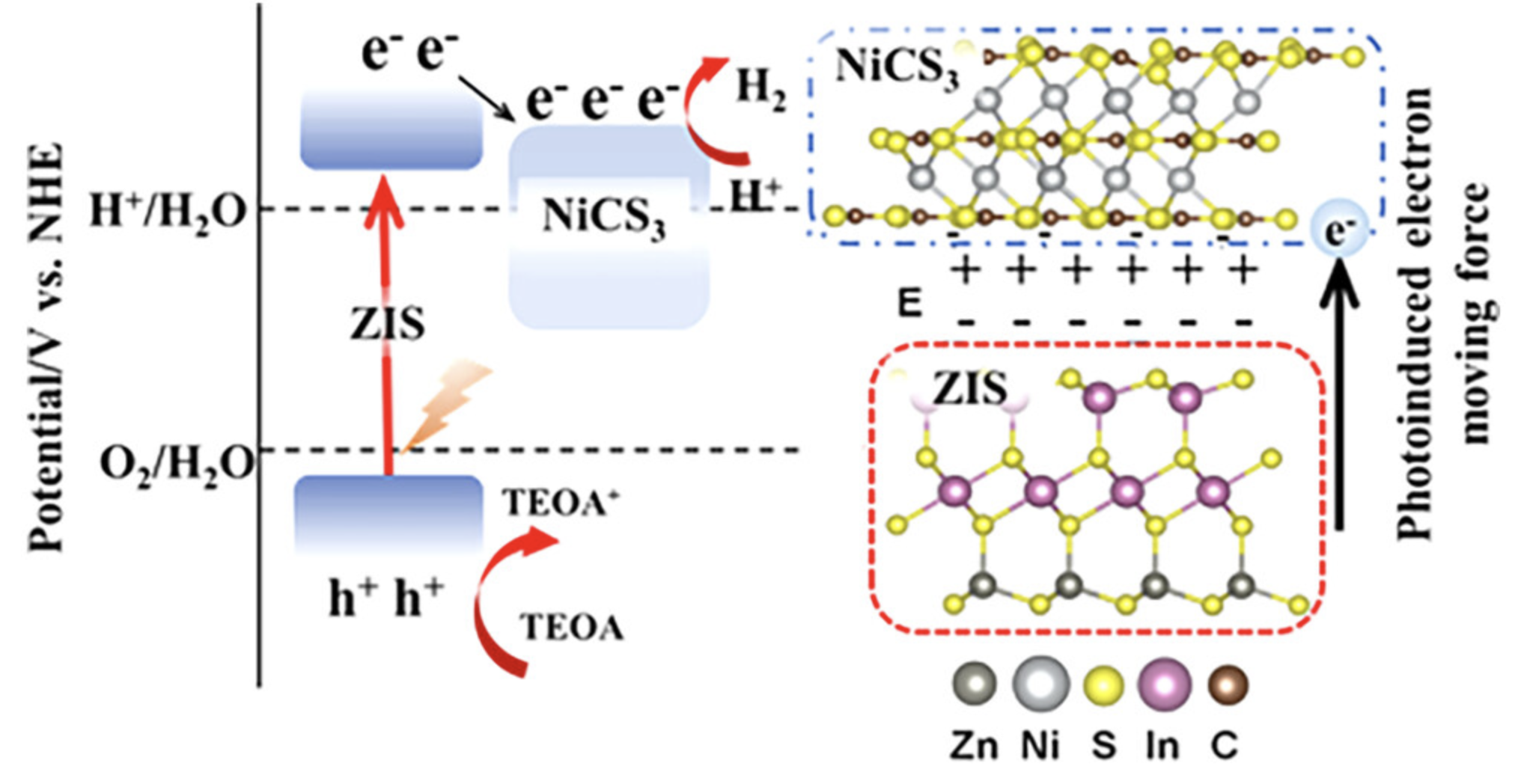
内建电场
影响电子和空穴在异质结中的输运。异质结内建电场是指在两种不同半导体材料接触时,由于能带差异而在界面附近形成的电场。
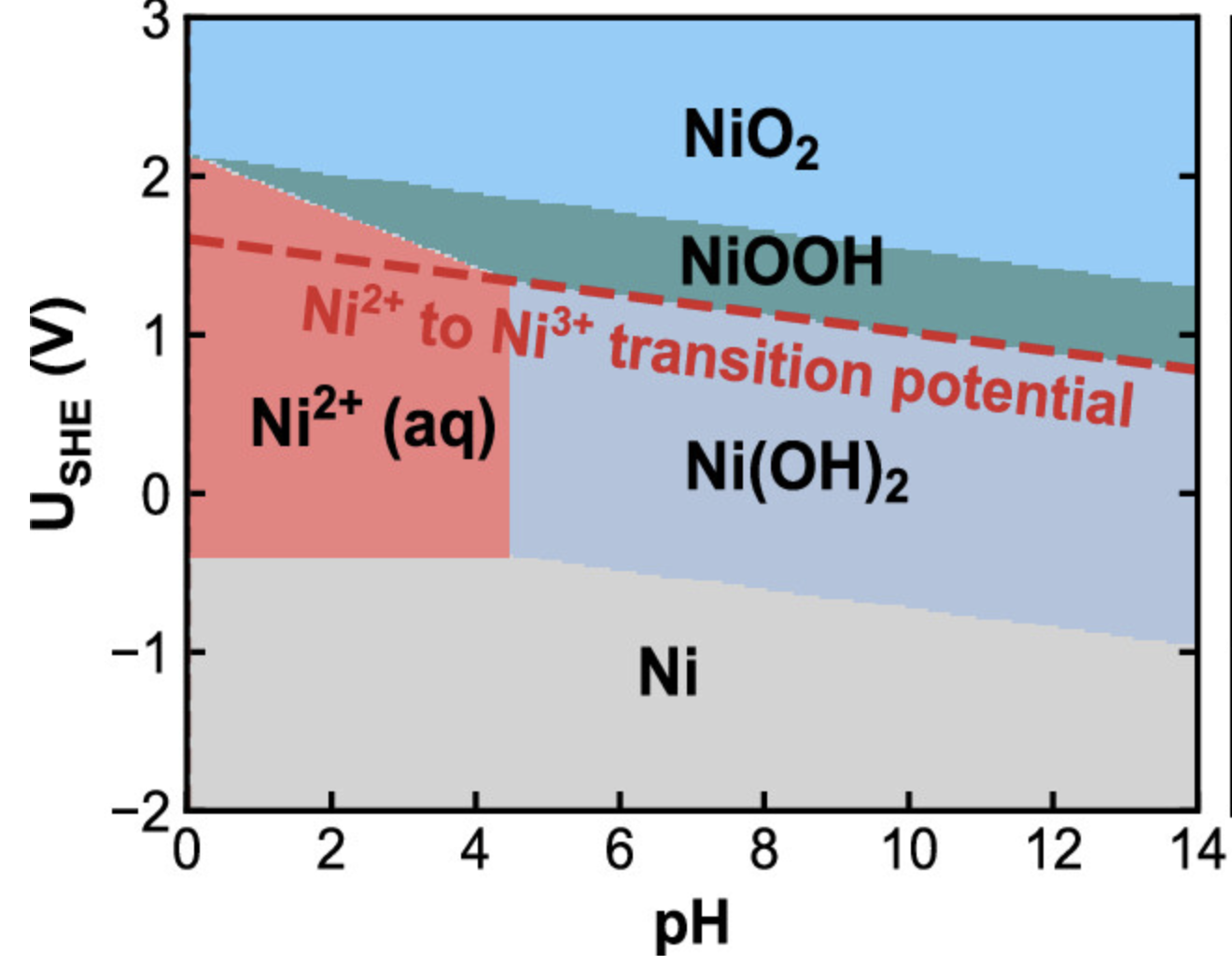
布拜图
判断在特定条件下哪种结构是稳定的。Pourbaix图,也称为电位-pH图或布拜图,是一种用于表示水溶液中不同氧化态的化学物质稳定性的图表。
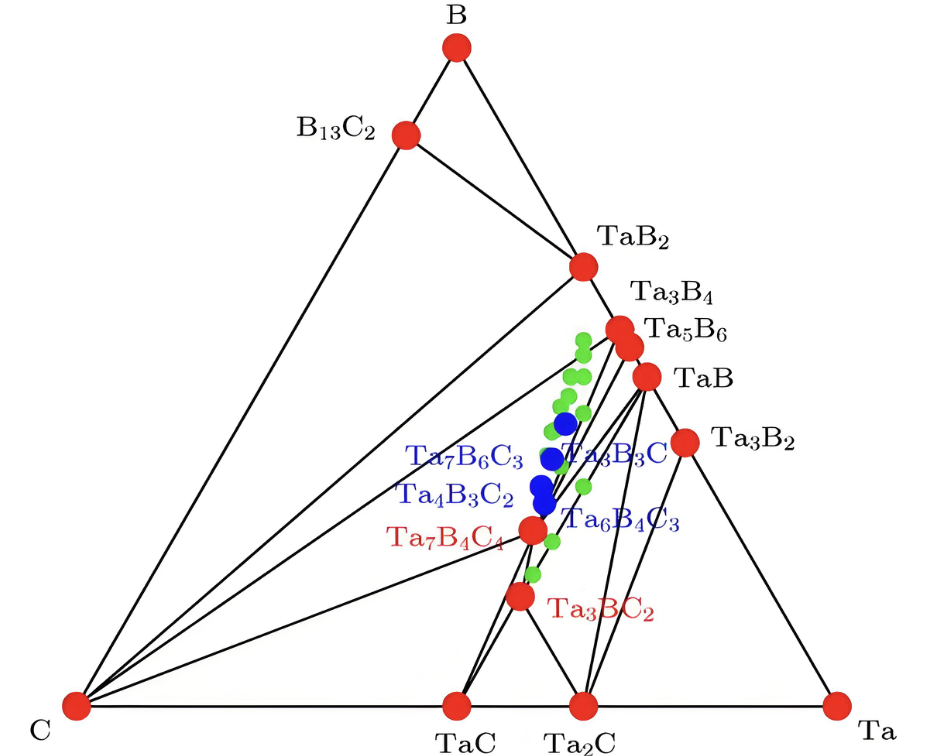
相图
了解材料在不同条件下的相变行为。结构的相图是材料科学中用来描述材料在不同条件(如温度、压力)下相变的图形表示。
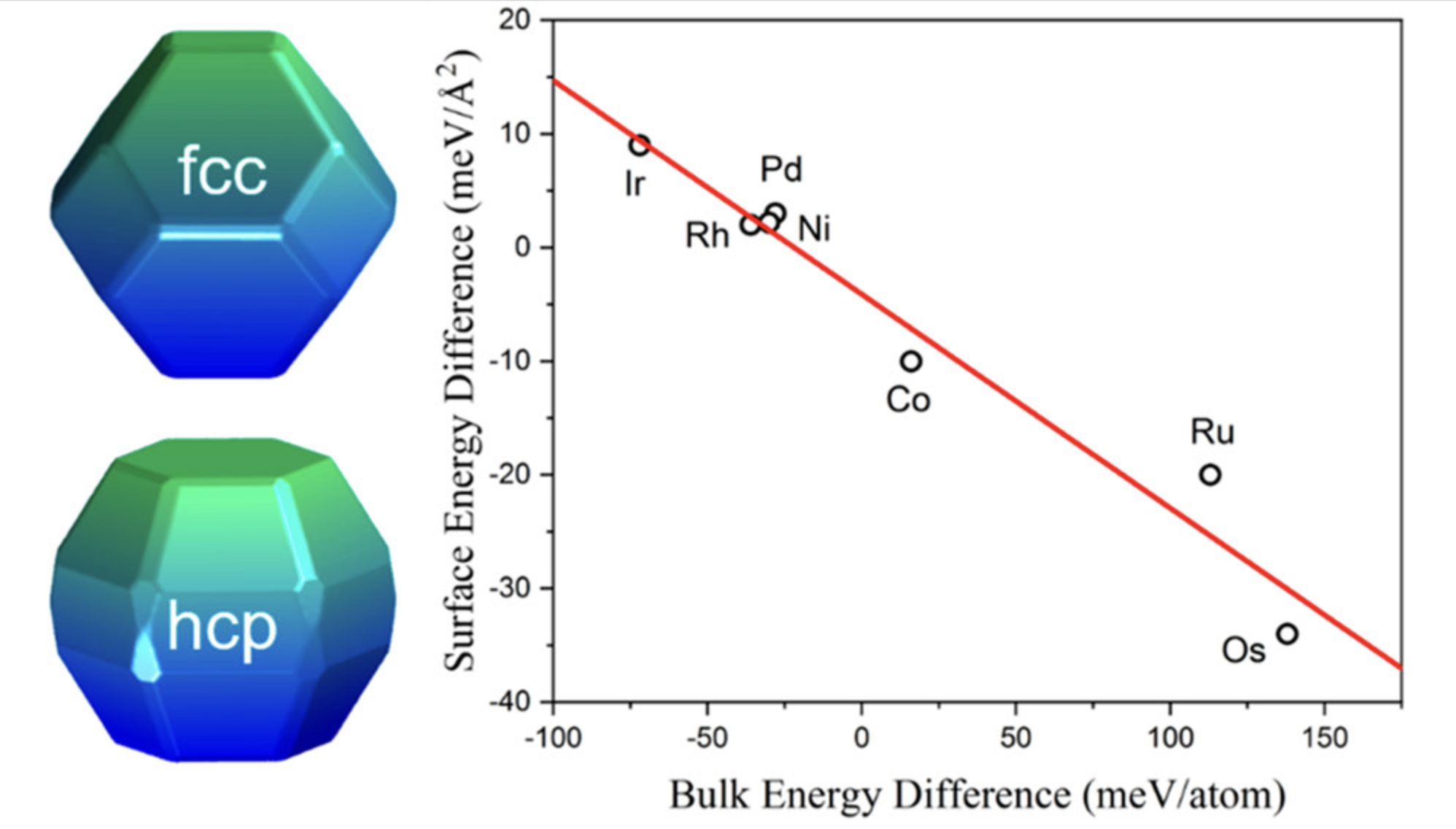
表面能
表面能越小,表面越容易形成。表面能可以理解为每单位面积产生一个新表面时所做的功。表面能不仅与表面的曲率有关,而且与表面原子排列的紧密度有关。
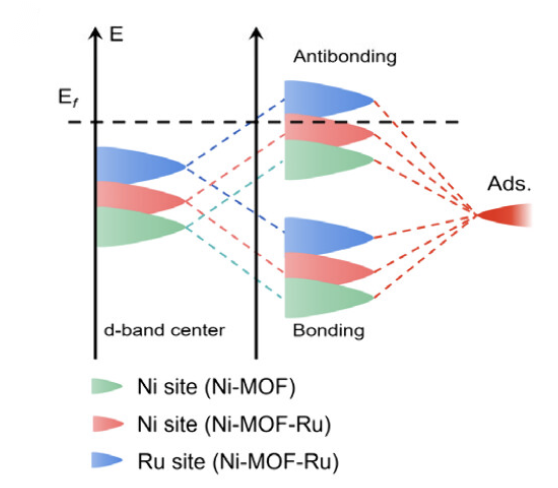
d带中心
离费米能级越近,吸附能力越强。金属催化剂的d带中心位置及其偏移是评价催化活性的一个重要参数。
静电势
体现了结构电子的分布情况。表面结构的静电势是指材料表面由于电子分布不均匀而产生的电势分布。
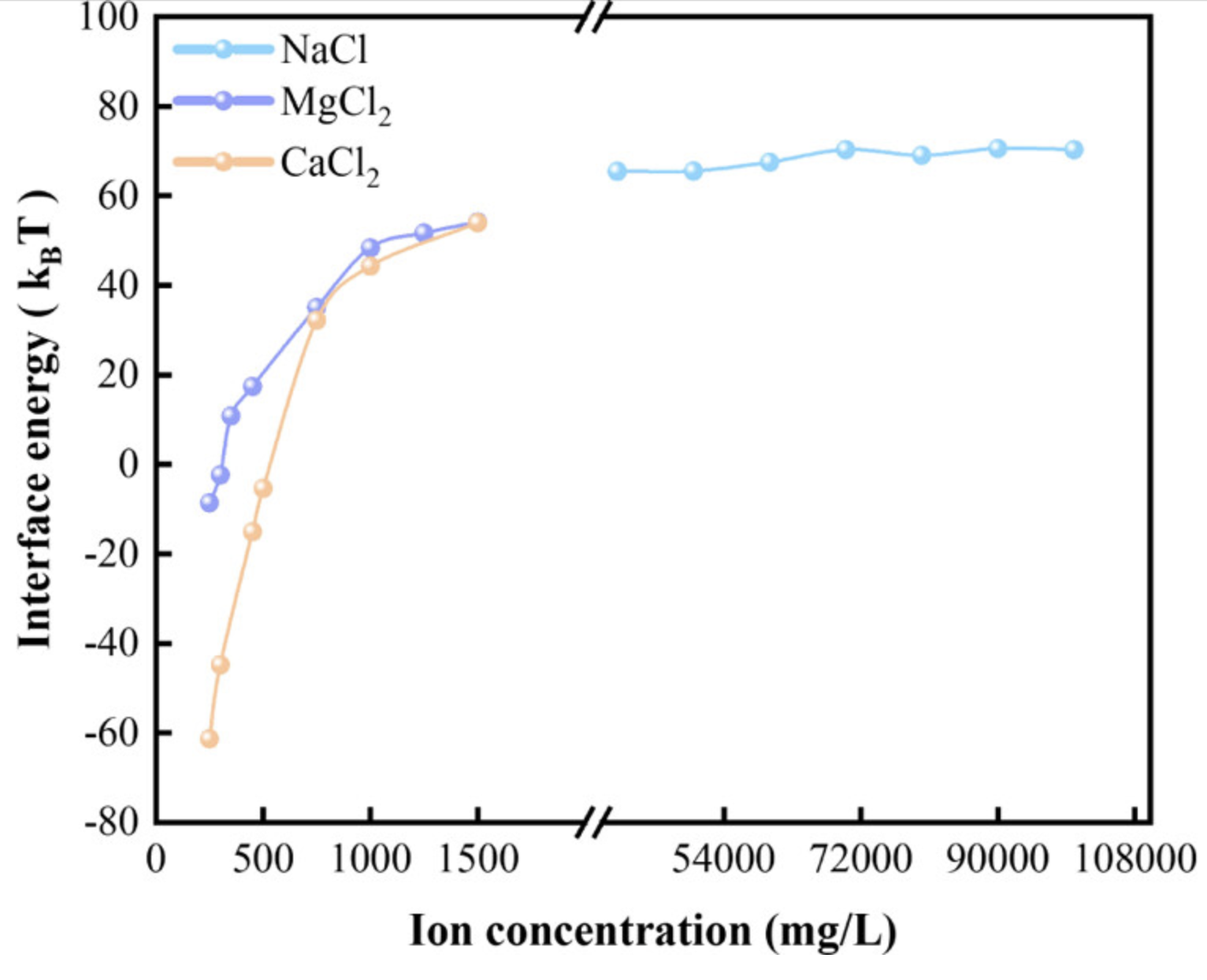
界面能
体现了形成界面的稳定性强弱。界面能是材料科学中一个重要的概念,它描述了两种不同材料界面上的相互作用能。
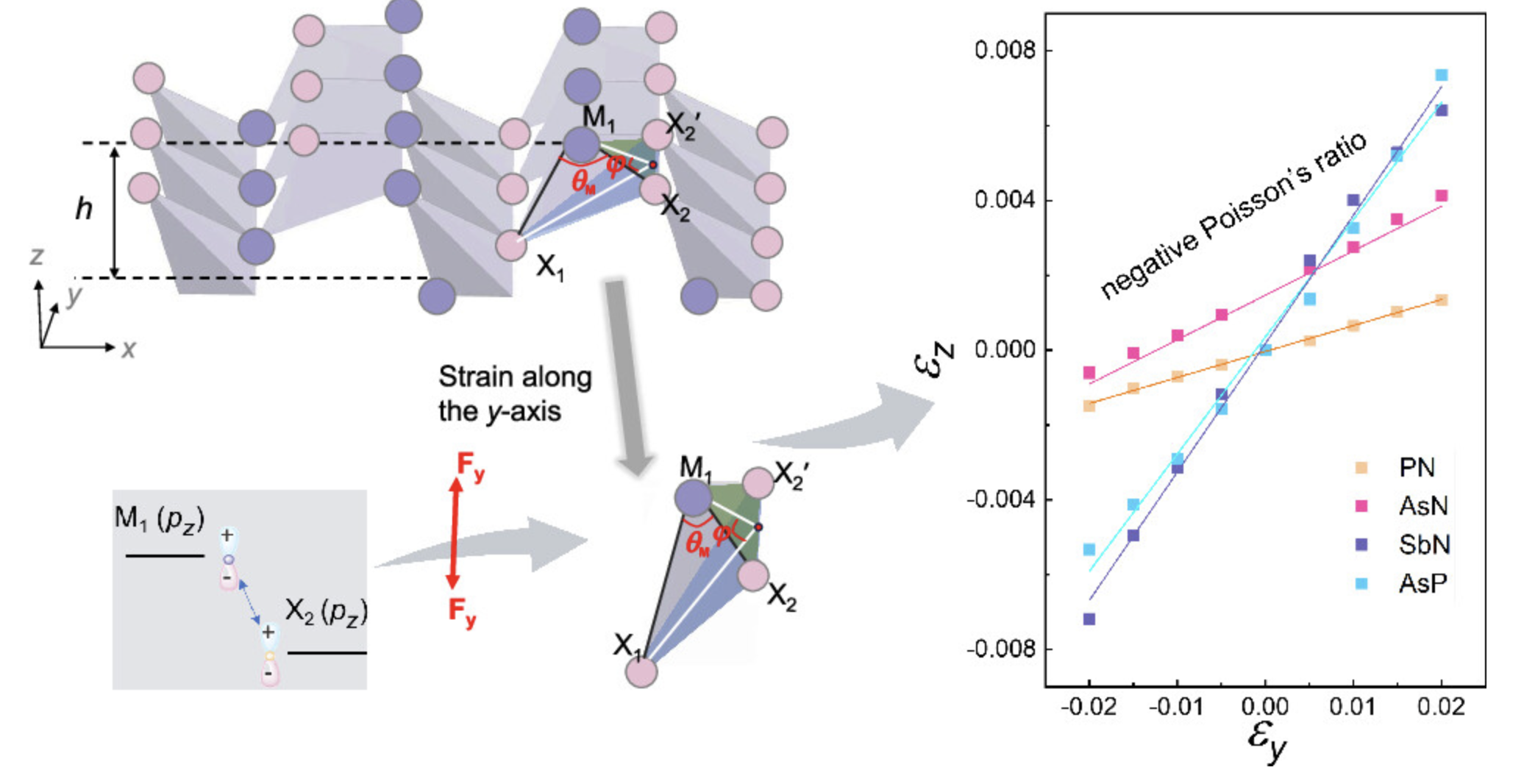
泊松比
材料在受到压缩或拉伸时的变形特性。泊松比是描述材料在受到压缩或拉伸时横向形变程度的无量纲物理量。
量子化学计算
基于量子力学原理的分子电子结构计算,提供精确的分子性质预测和反应机理研究
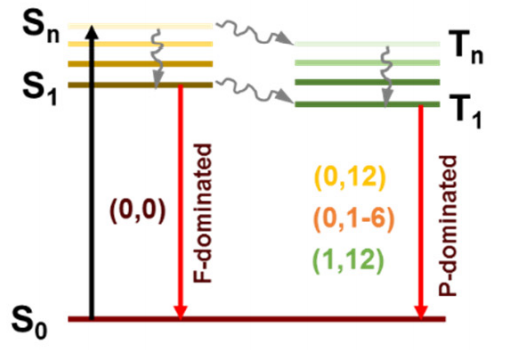
激发态
计算分子的电子激发态,分析光吸收、发射光谱和光化学反应机理。
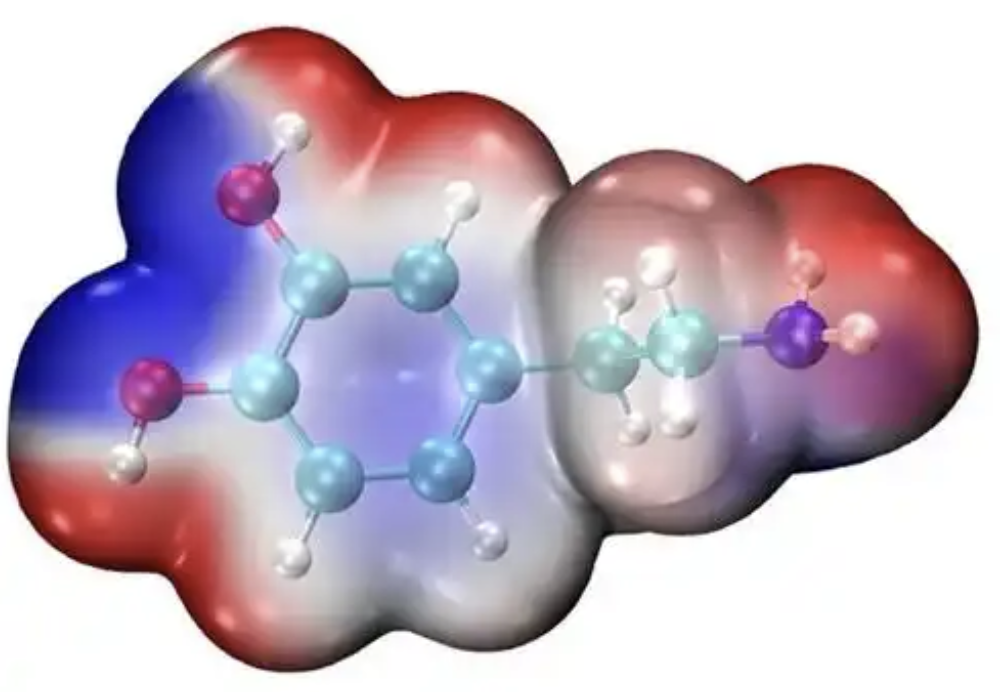
静电势
计算分子表面的静电势分布,预测分子间相互作用和反应活性位点。
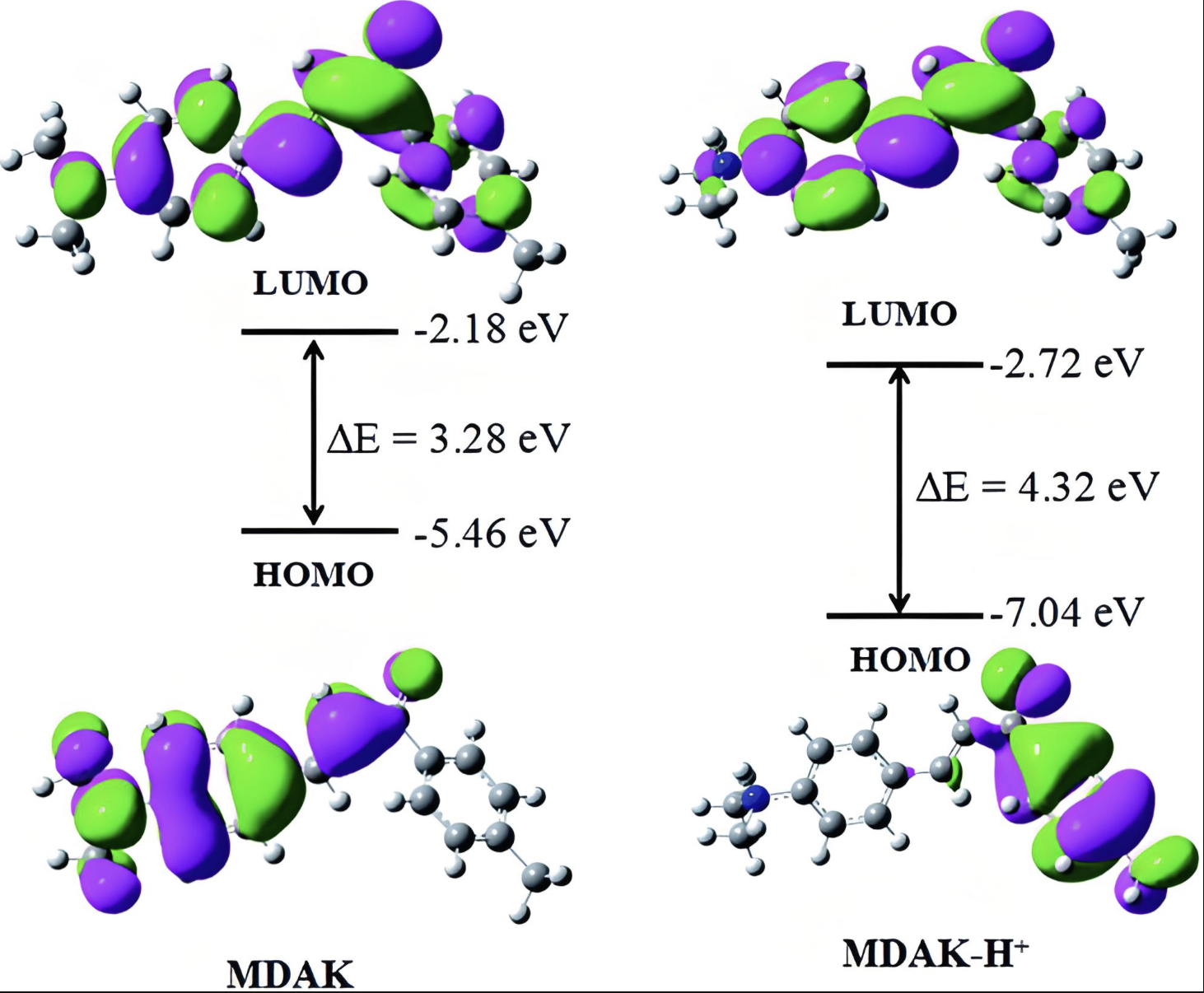
HOMO-LUMO
分析最高占据分子轨道和最低未占据分子轨道,预测化学反应活性。
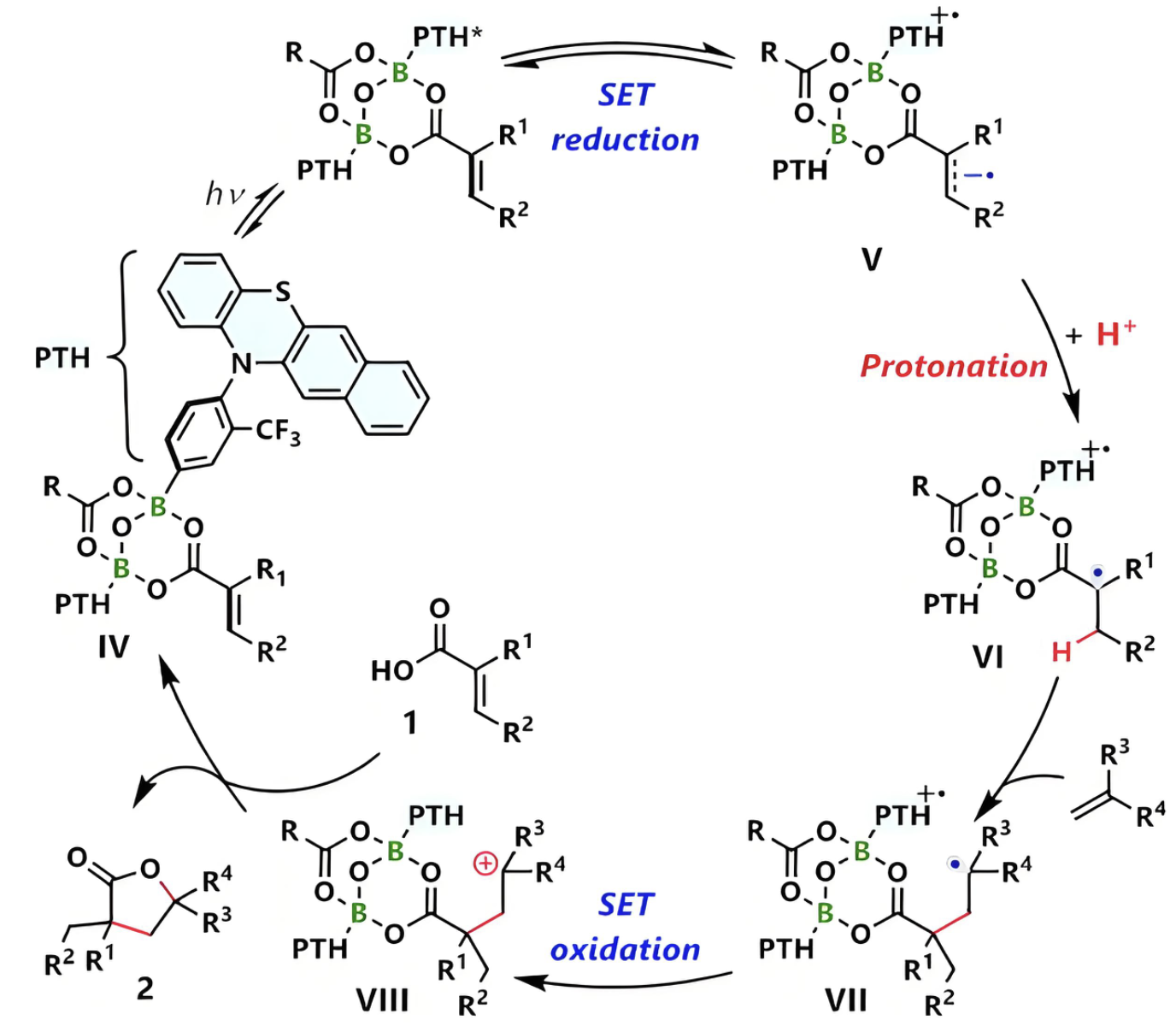
反应路径
计算化学反应的完整路径,包括反应物、过渡态和产物的优化结构。
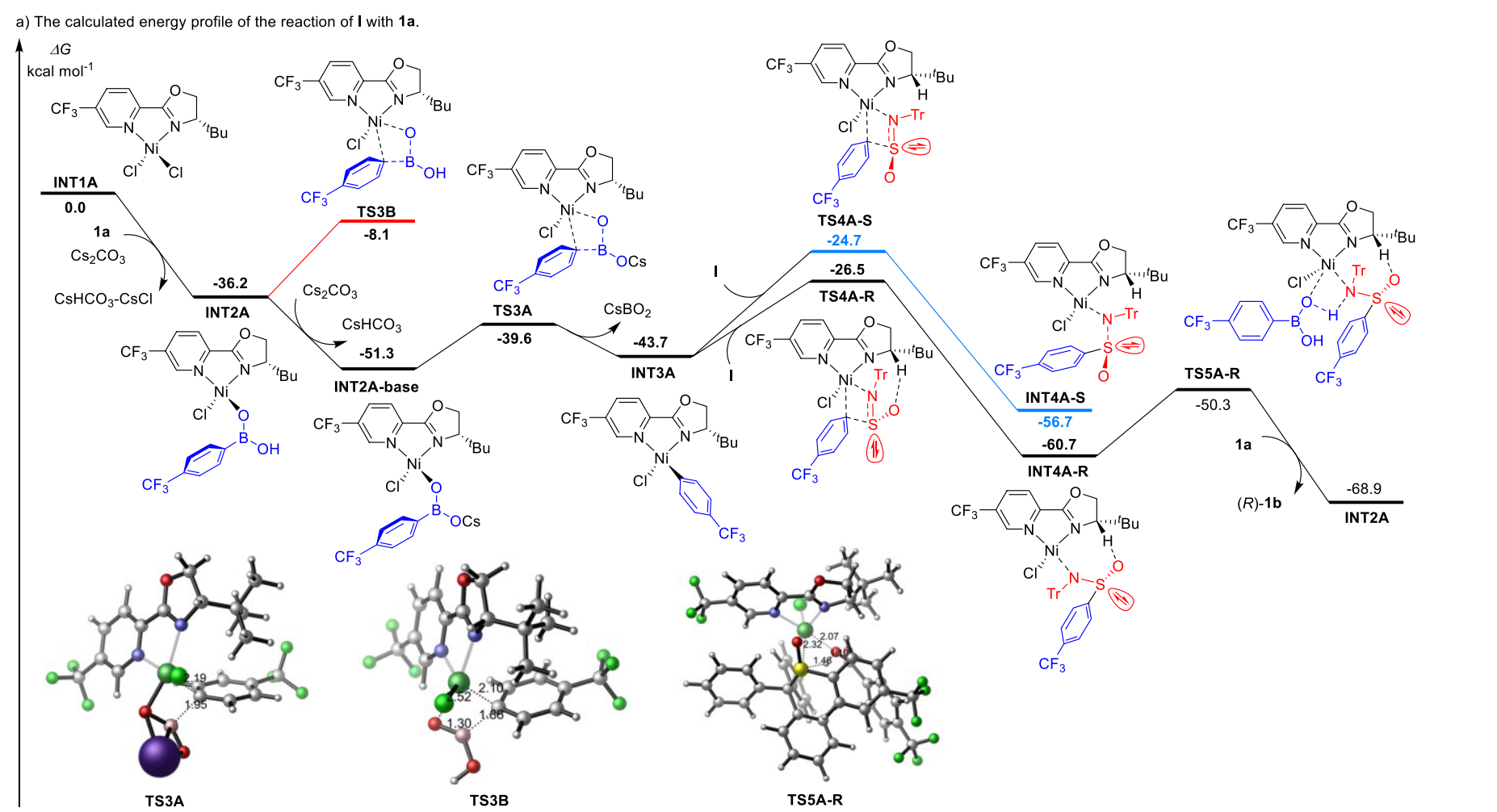
过渡态
定位化学反应的过渡态结构,计算活化能和反应速率常数。
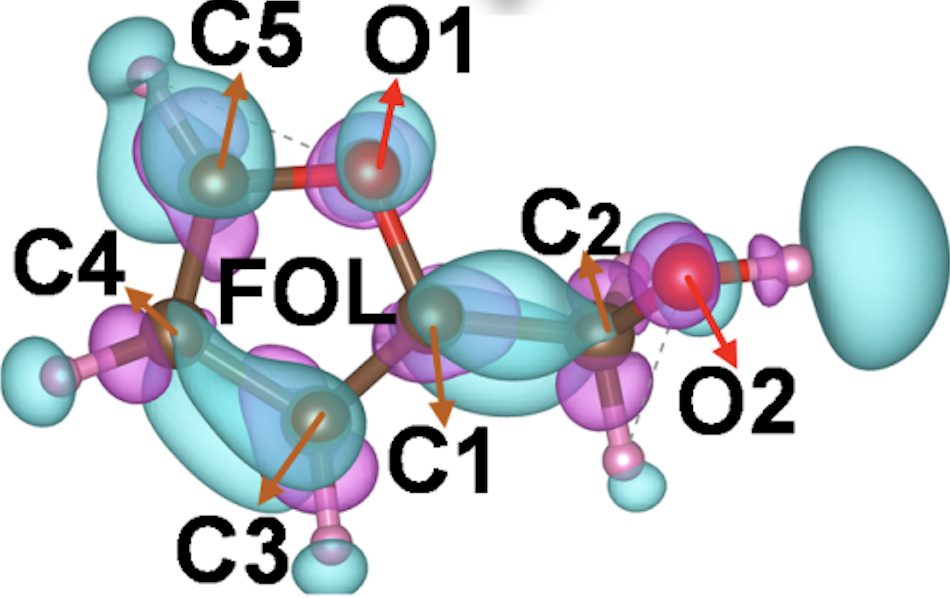
福井函数
计算分子的福井函数,预测亲电和亲核反应的活性位点。
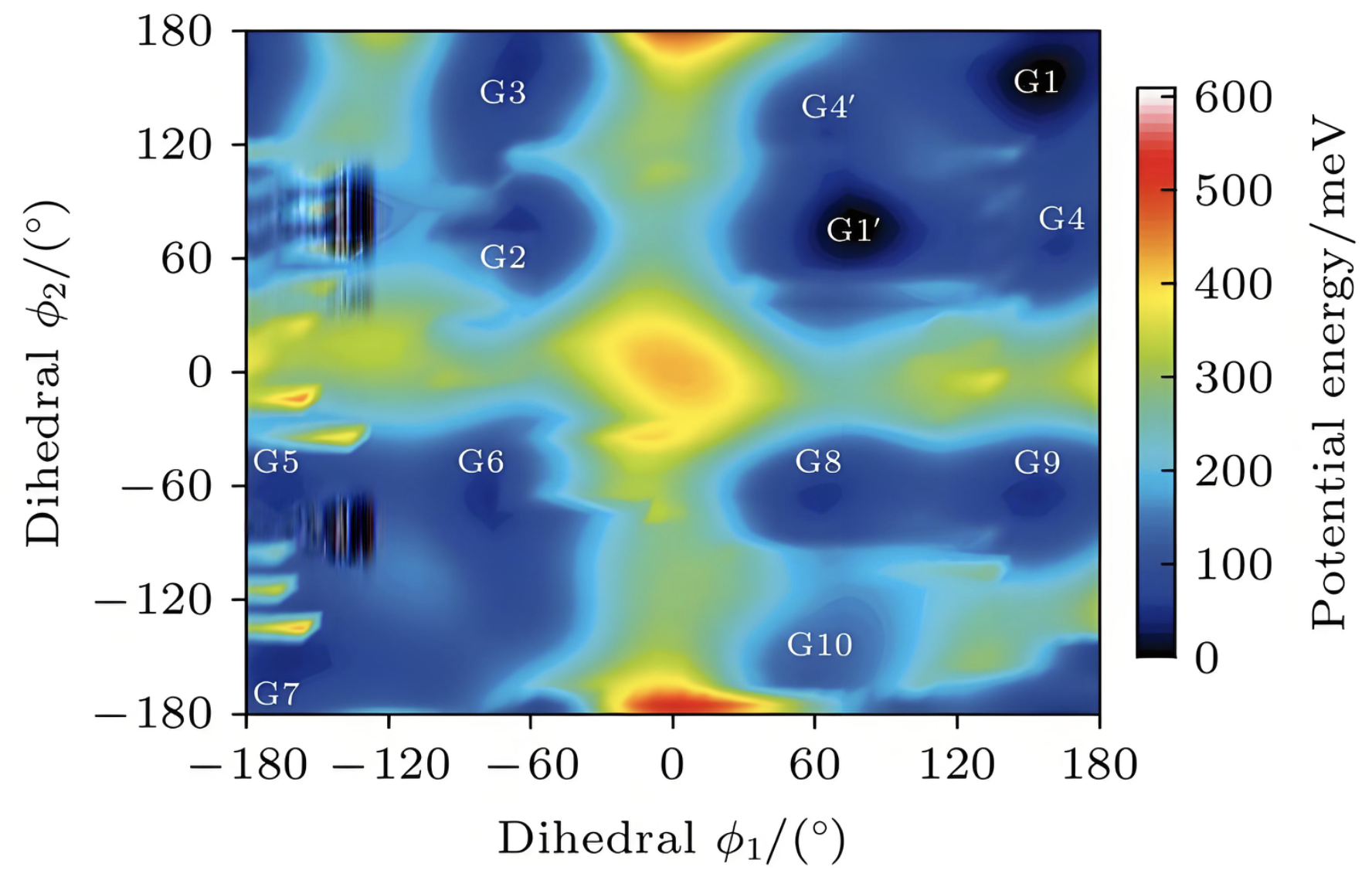
势能面扫描
扫描分子构象变化的势能面,寻找最稳定构象和反应路径。
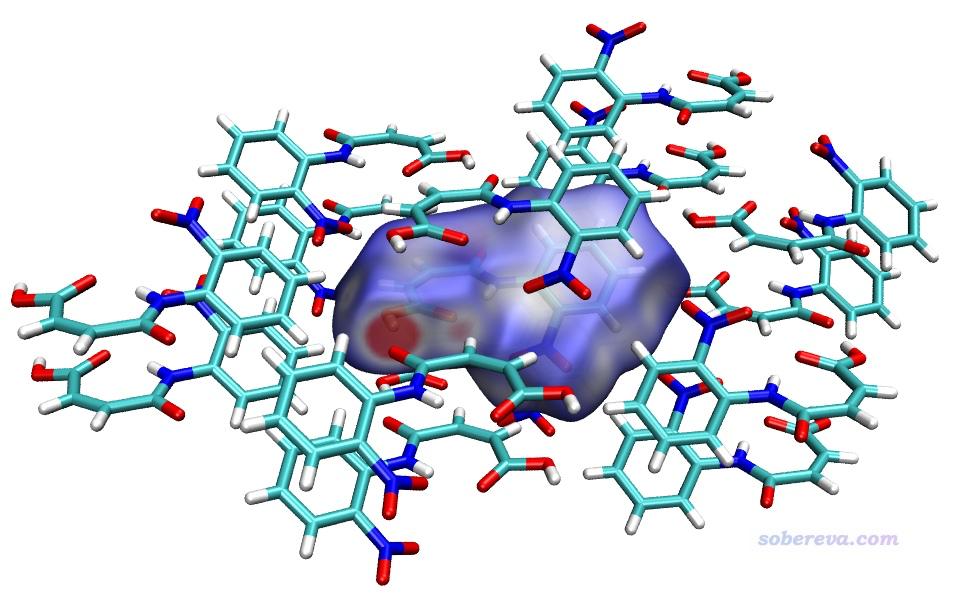
Hirshfeld表面分析
分析分子间相互作用的三维可视化,量化氢键、范德华力等弱相互作用。
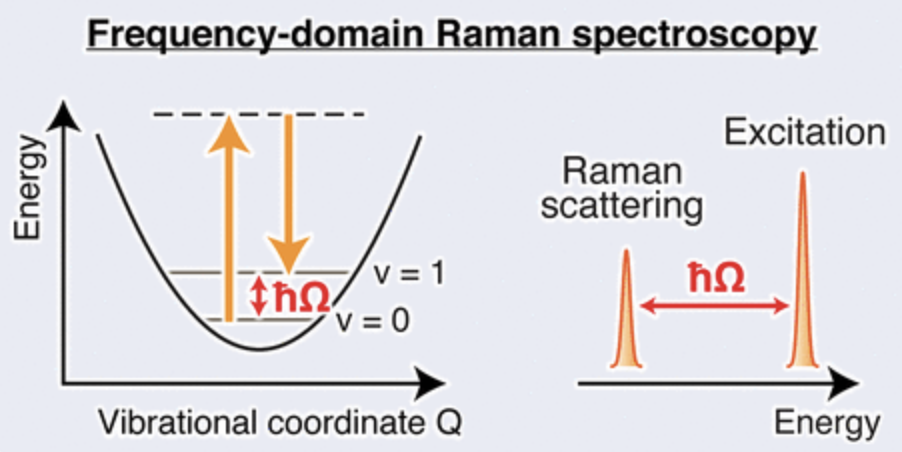
拉曼光谱
计算分子的拉曼散射光谱,辅助光谱归属和分子结构分析。
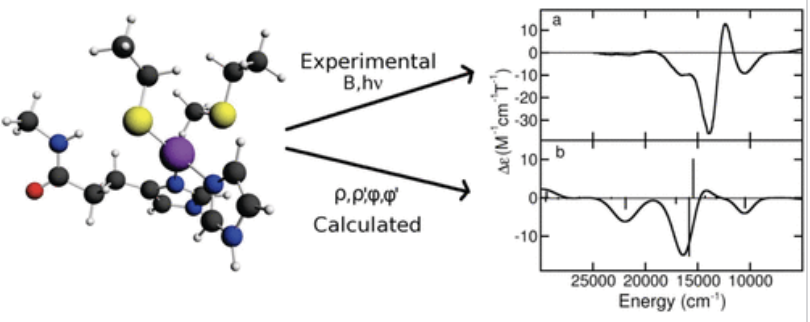
圆二色谱
计算手性分子的圆二色性光谱,分析分子的立体化学结构。
偶极矩
计算分子的电偶极矩、磁偶极矩和极化率,预测分子的极性。
结合能
计算分子间复合物的结合能,分析超分子相互作用强度。
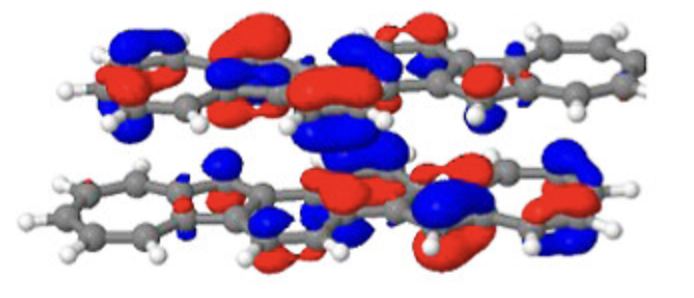
差分电荷密度
分析化学键形成过程中的电荷重新分布和电荷转移。
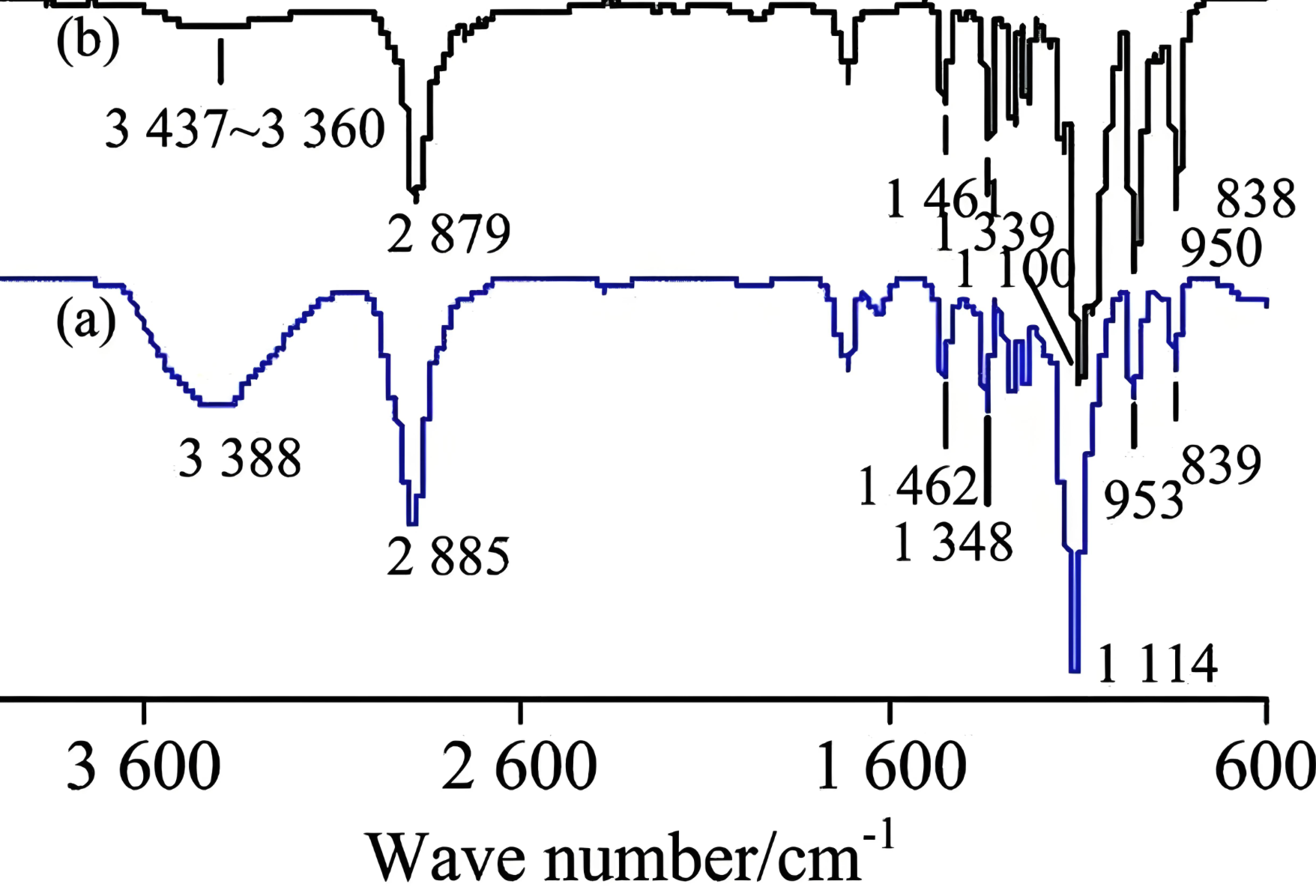
红外光谱
计算分子的红外振动光谱,辅助实验光谱归属和结构确认。
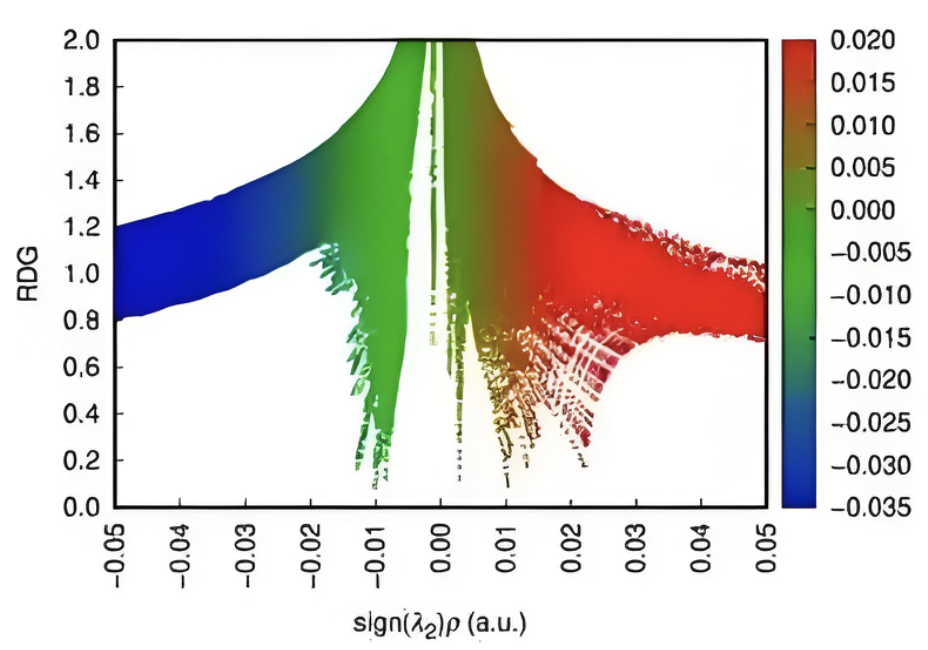
独立梯度模型
分析弱相互作用的本质和强度,可视化氢键、范德华力等。
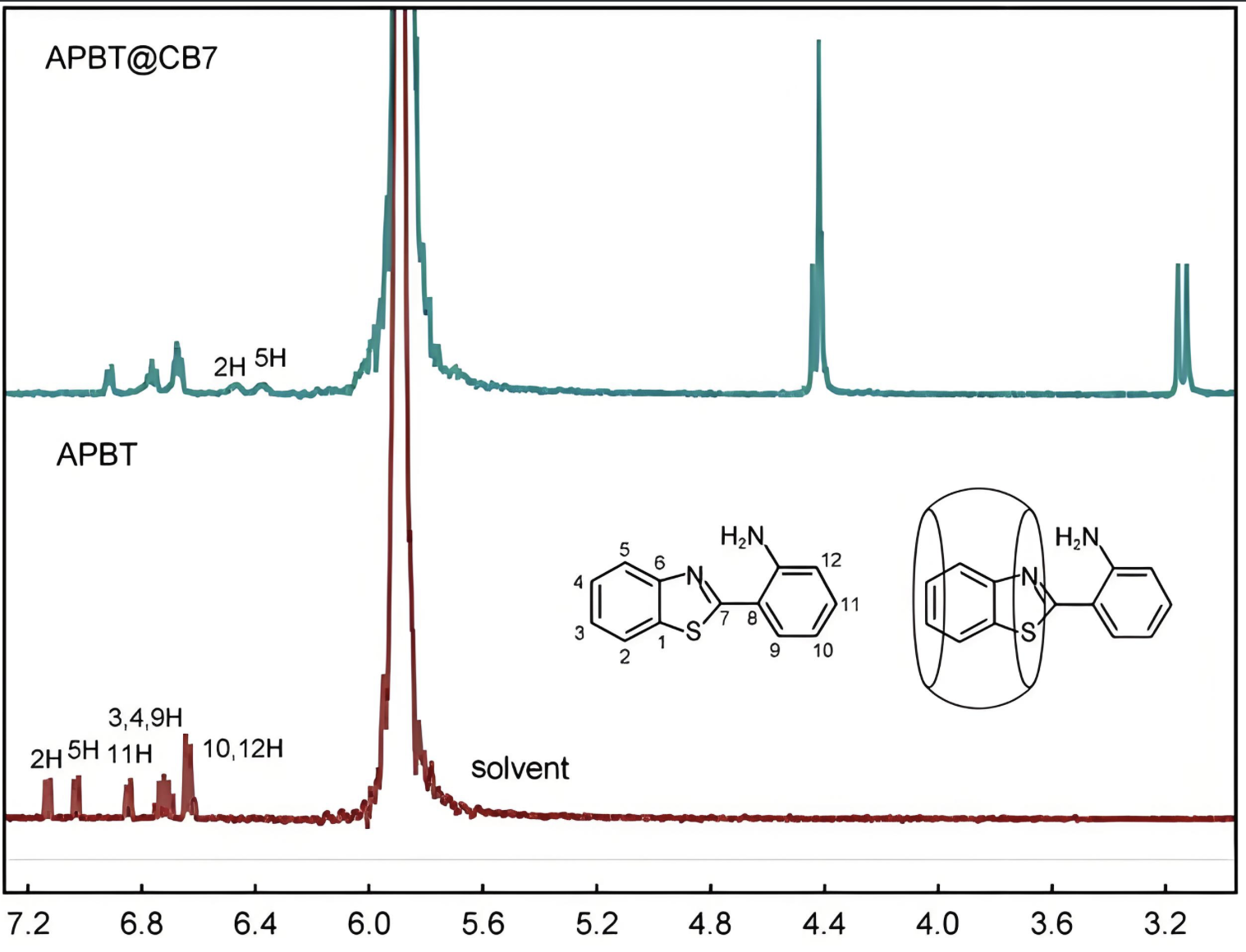
核磁共振谱
计算分子的NMR化学位移和耦合常数,辅助结构解析。
形成焓
计算分子或反应的标准形成焓,评估热力学稳定性。
紫外可见光谱
计算分子的UV-Vis吸收光谱,预测光学性质和激发态性质。
自旋密度
分析自由基和过渡金属配合物的自旋密度分布。
氢键
分析分子内和分子间氢键的几何参数和相互作用能。
芳香性分析
计算分子的芳香性指数和芳香性电流密度,评估芳香稳定性。
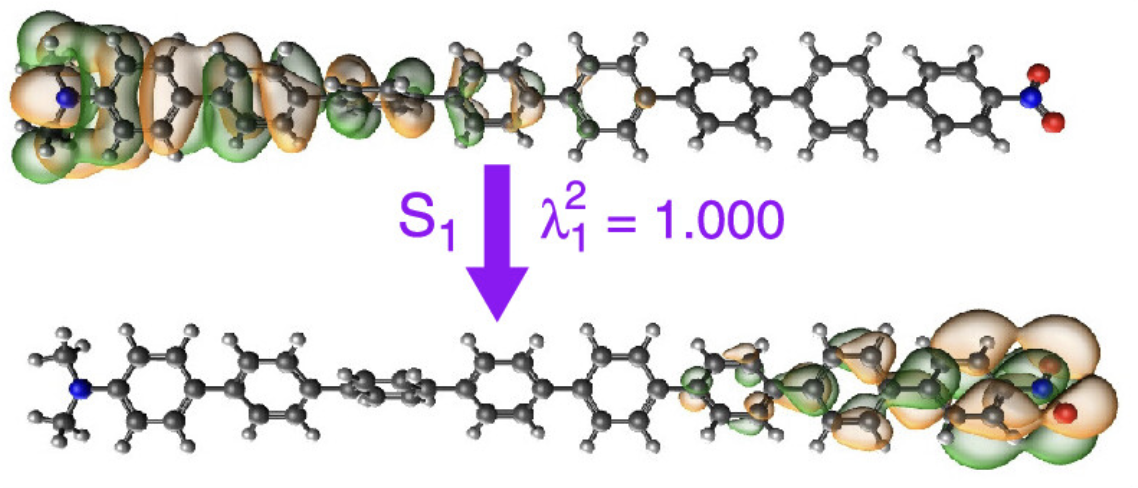
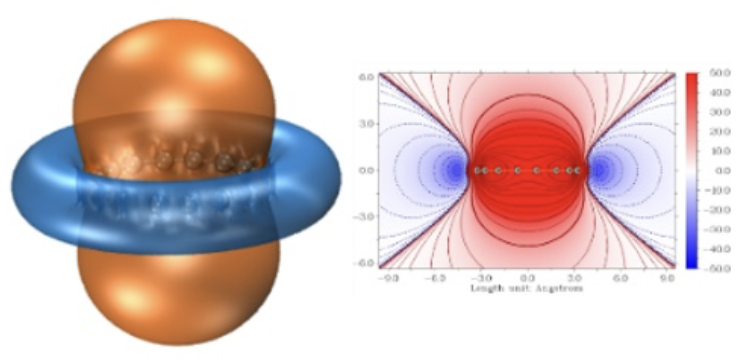
空穴-电子分析
分析激发态的空穴-电子分布,理解电荷转移特性。
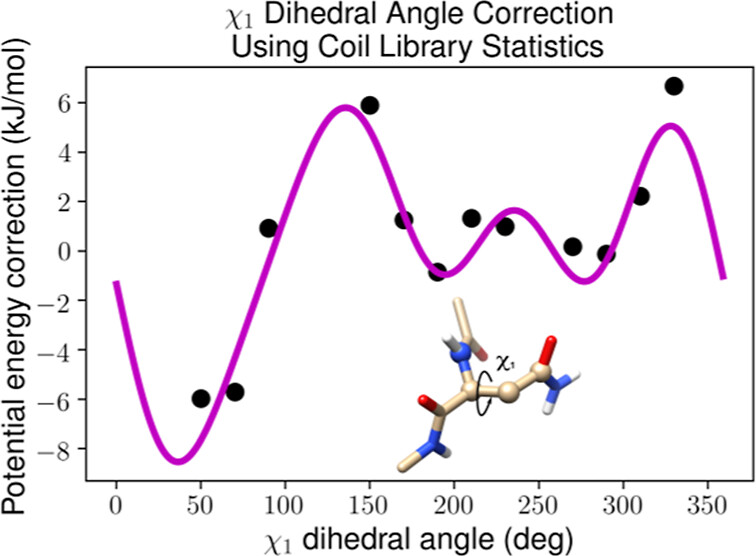
二面角
分析分子构象中关键二面角的变化,优化分子几何结构。

磁感应电流密度
计算分子在磁场中的感应电流密度,分析芳香性和反芳香性。
分子动力学模拟
基于经典力学的分子运动模拟,研究材料的动态行为和热力学性质
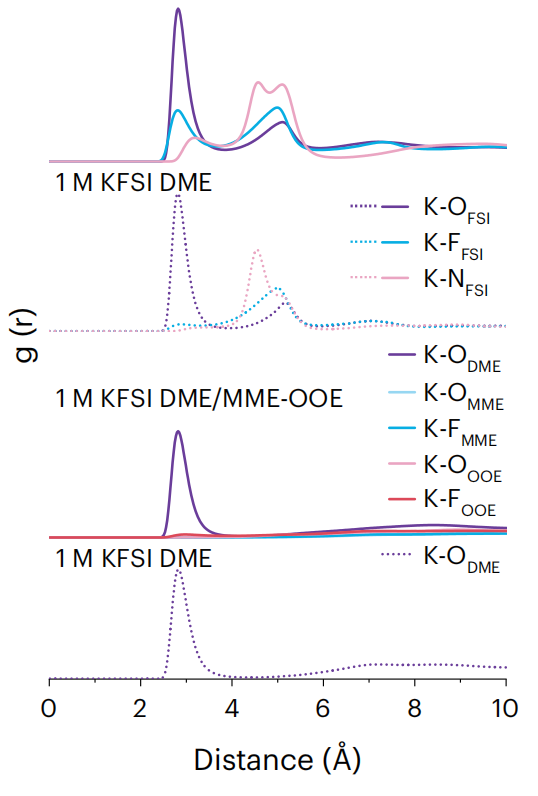
径向分布函数
分析原子间的空间分布规律,揭示材料的短程有序结构特征。
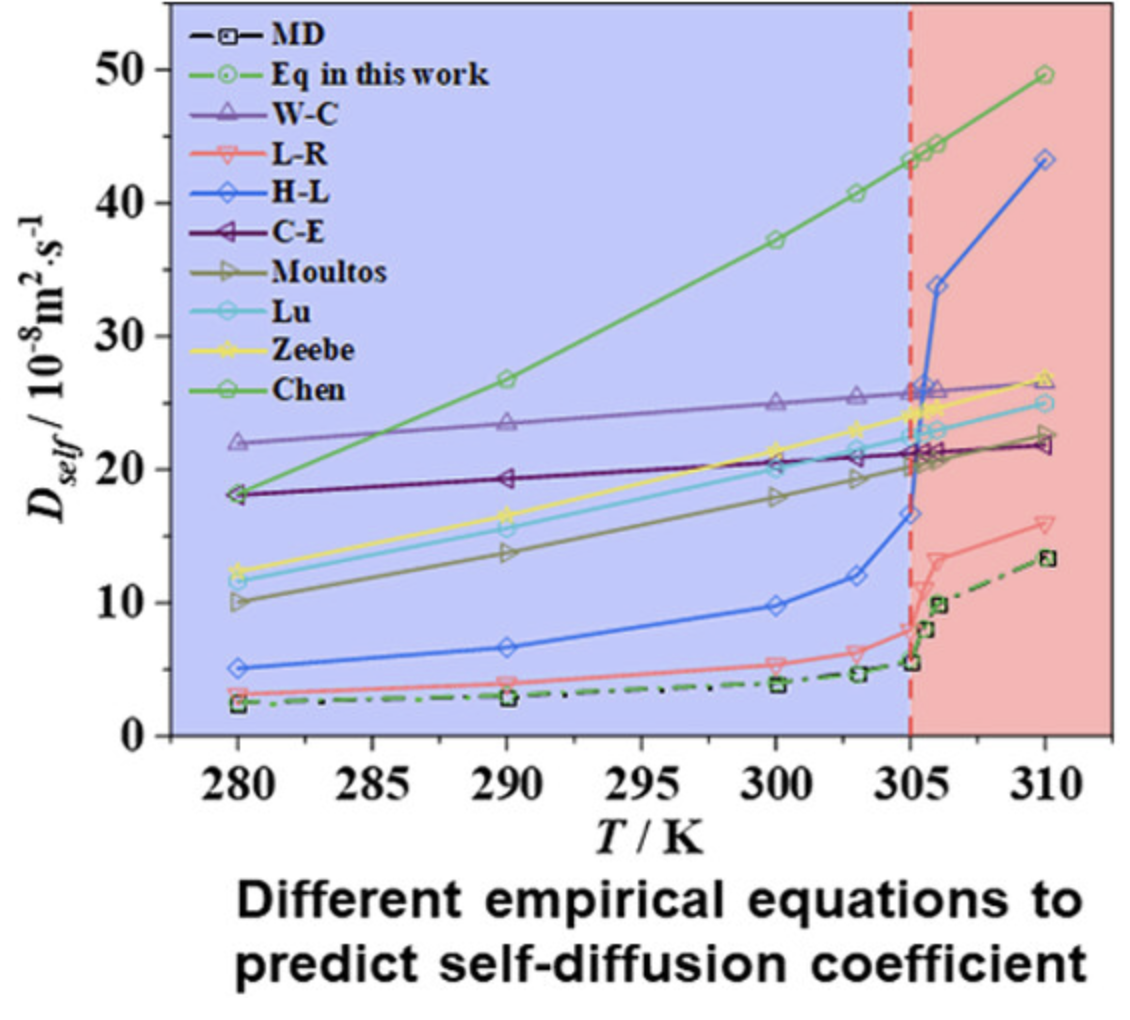
扩散系数
计算离子或分子在材料中的扩散能力,基于均方位移计算。
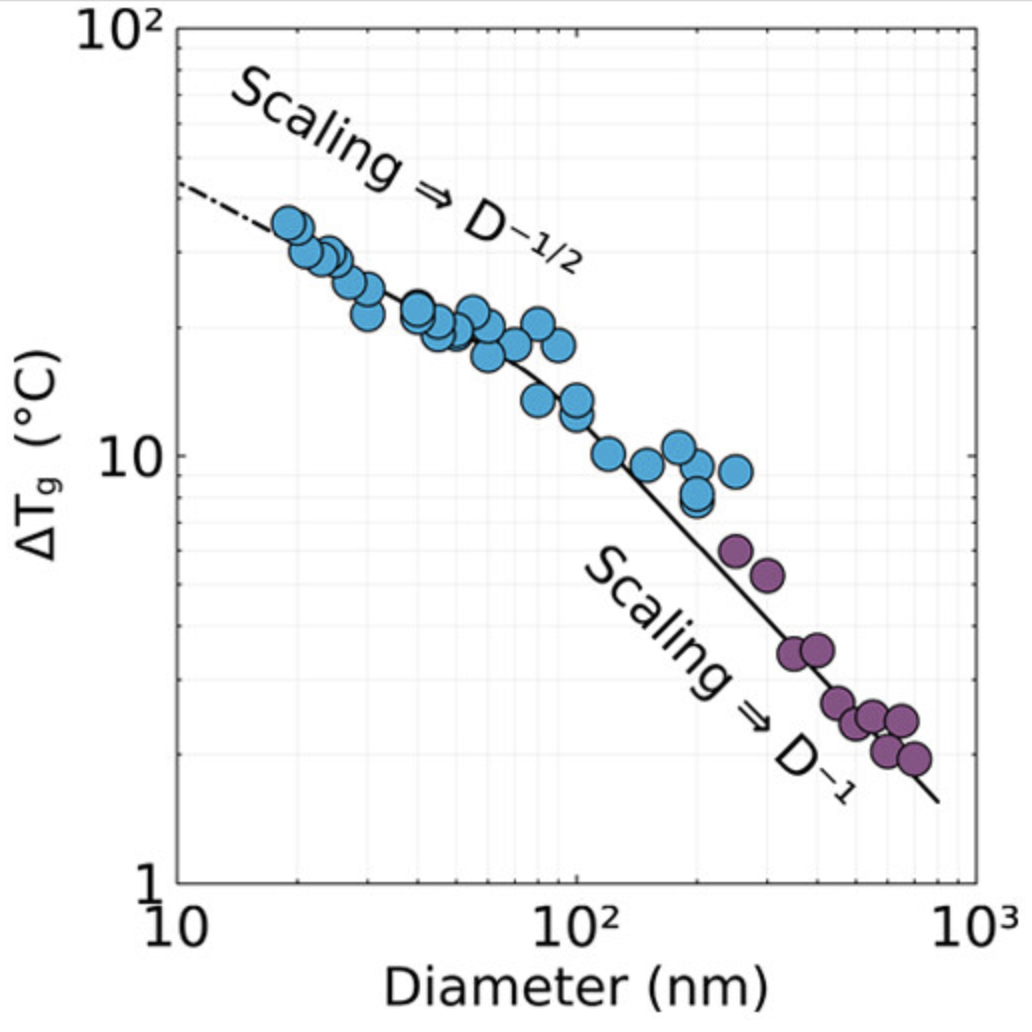
玻璃化转变温度
计算聚合物材料的玻璃化转变温度,评估材料的热性能。

电池电解液
模拟电解液中离子传导行为,优化电池电解液配方和性能。
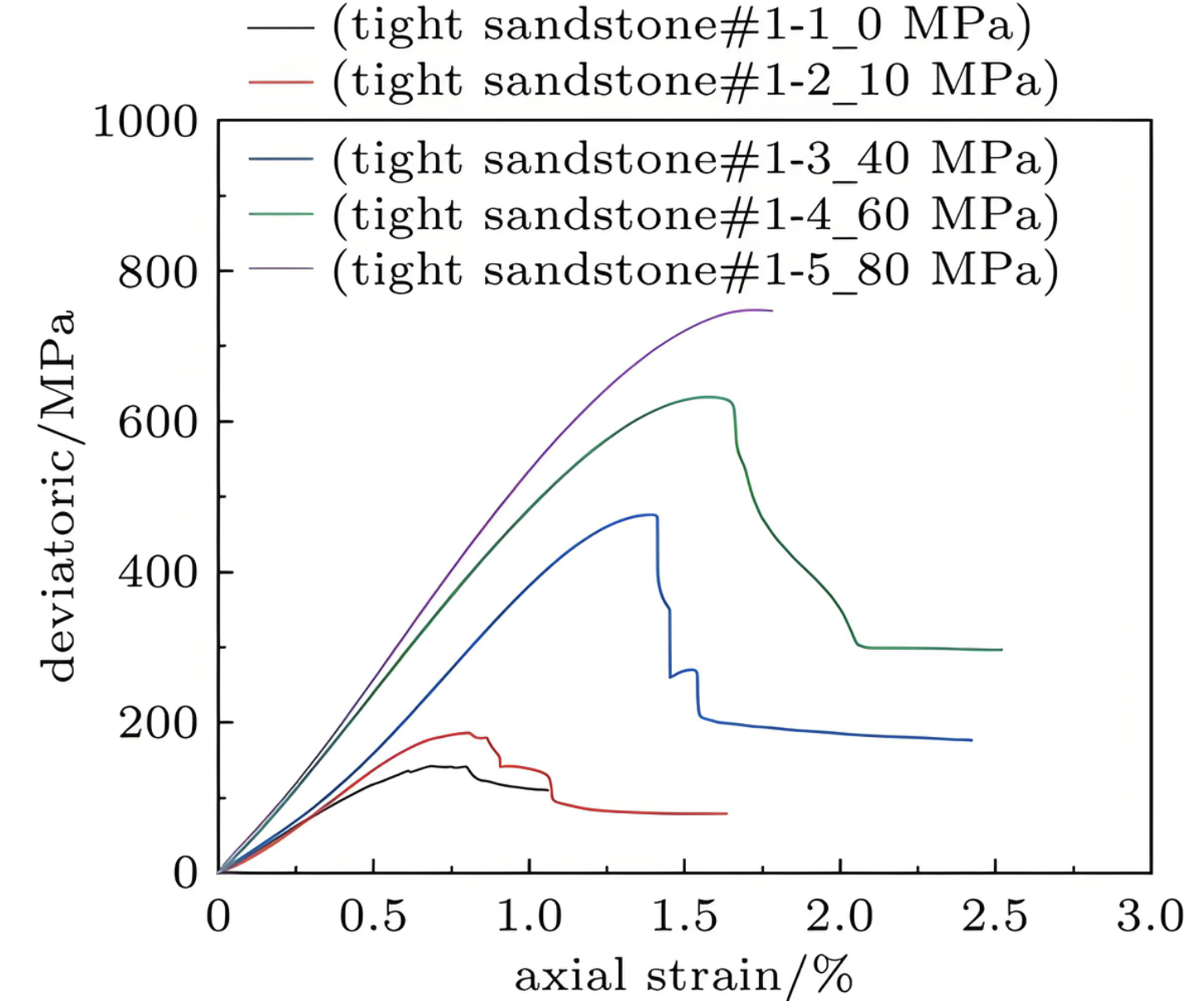
应力应变曲线
计算材料在机械载荷下的力学响应,获得弹性模量等力学参数。
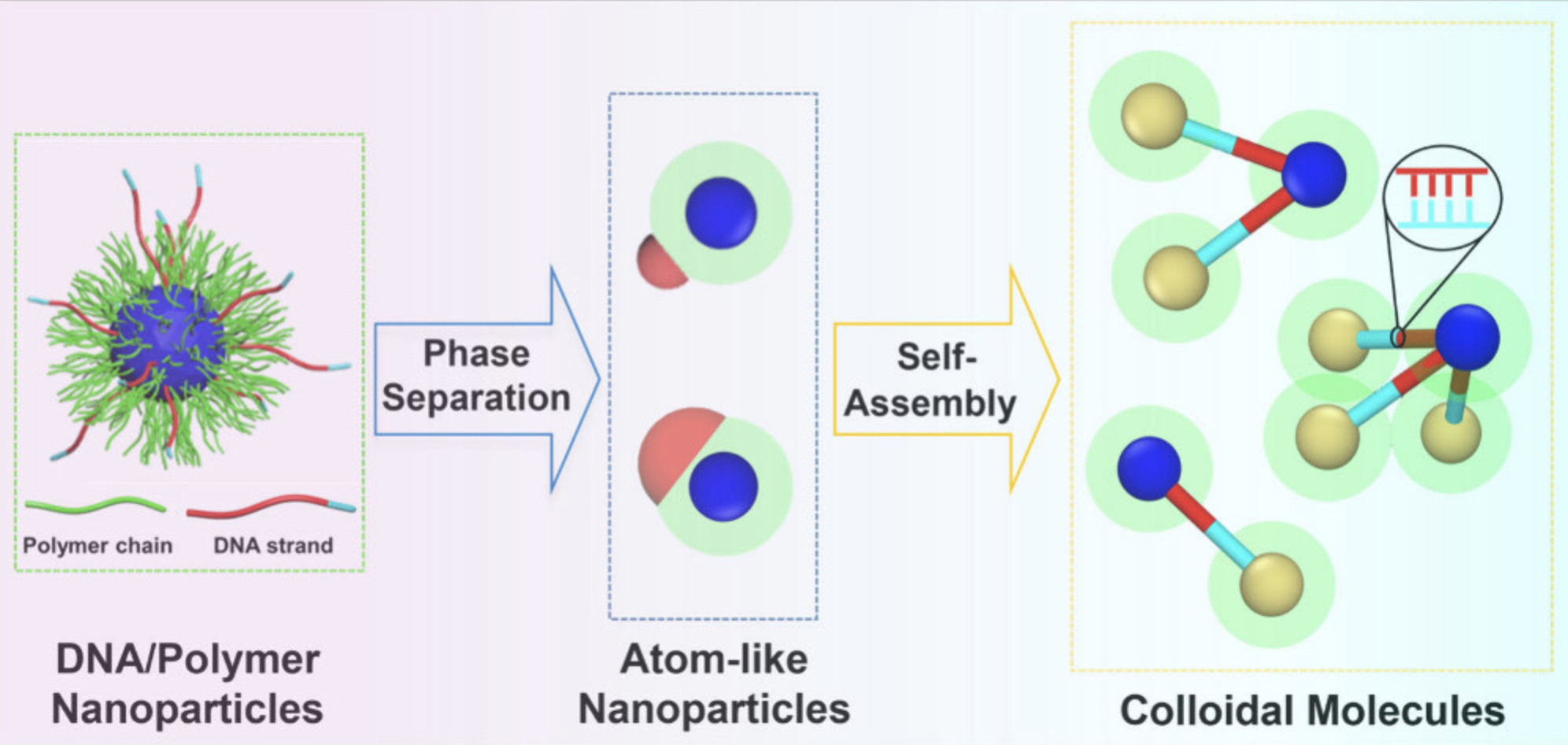
自组装
模拟分子或纳米颗粒的自组装过程,研究有序结构的形成机理。
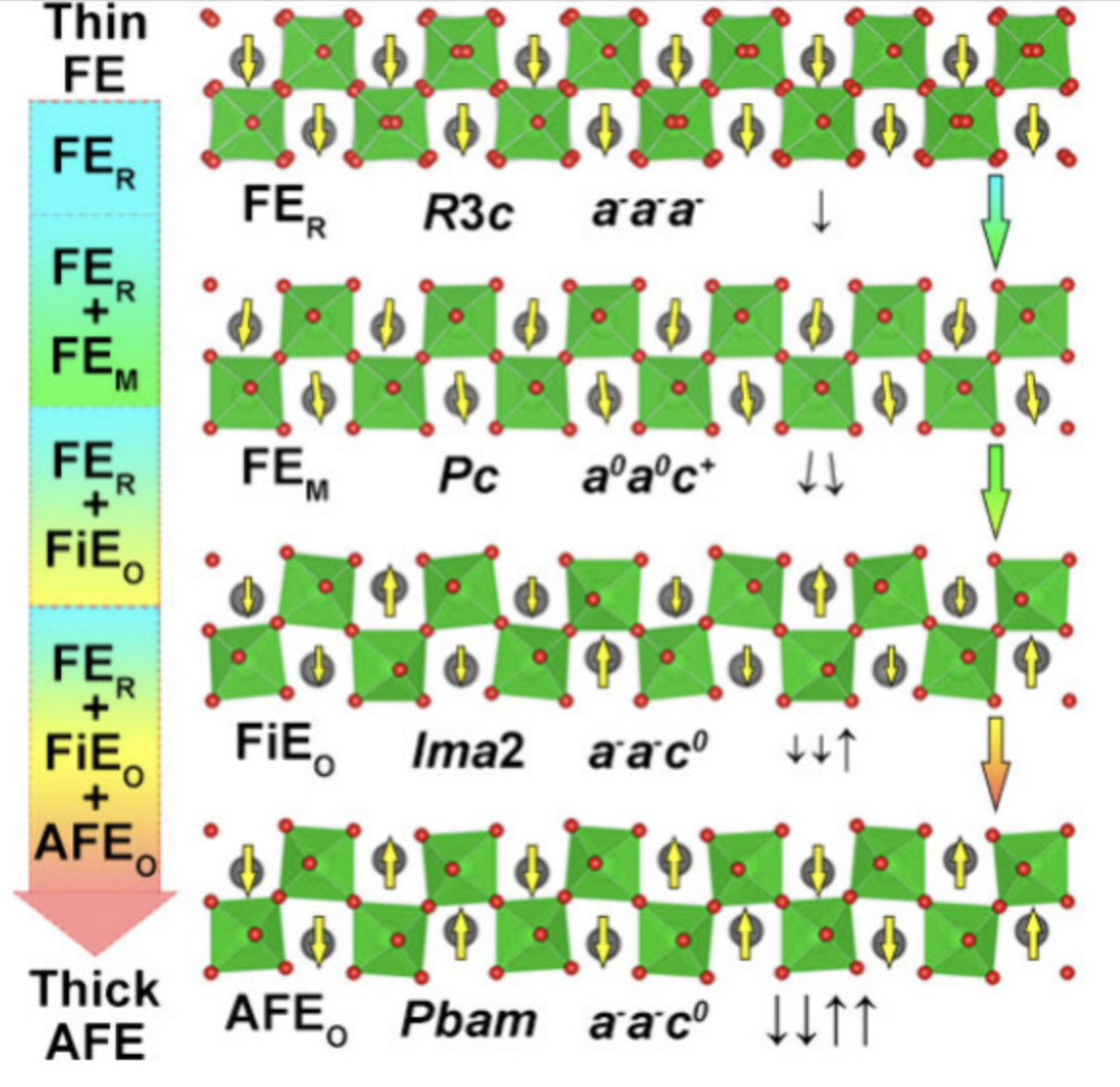
相转变
研究材料在不同条件下的相变行为,包括熔化、结晶等过程。
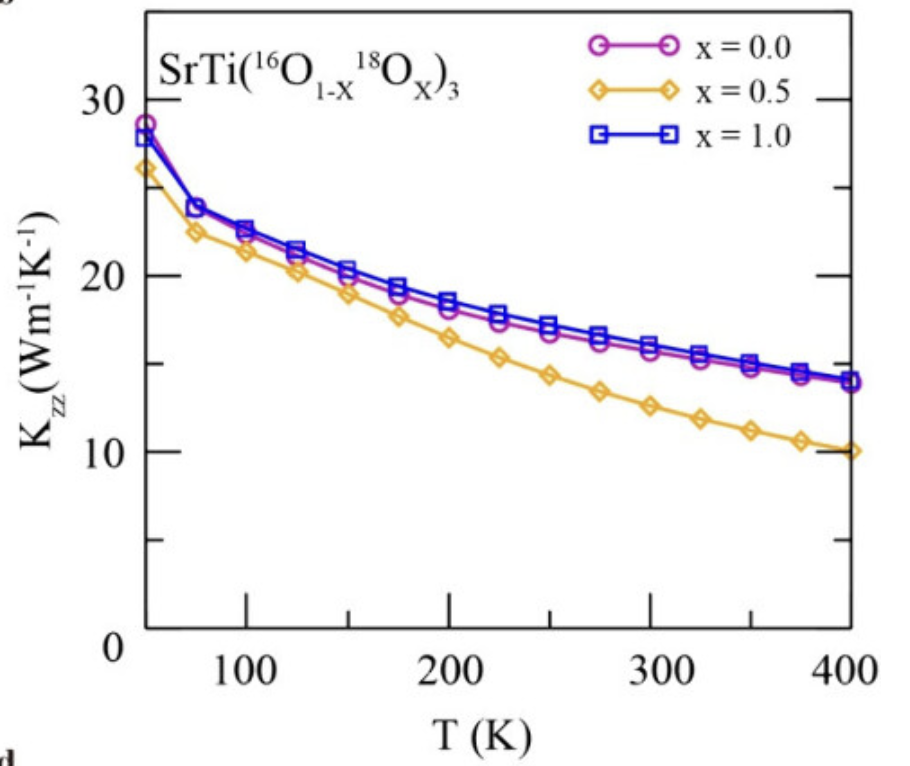
热导率
计算材料的热传导性能,基于Green-Kubo公式或非平衡方法。
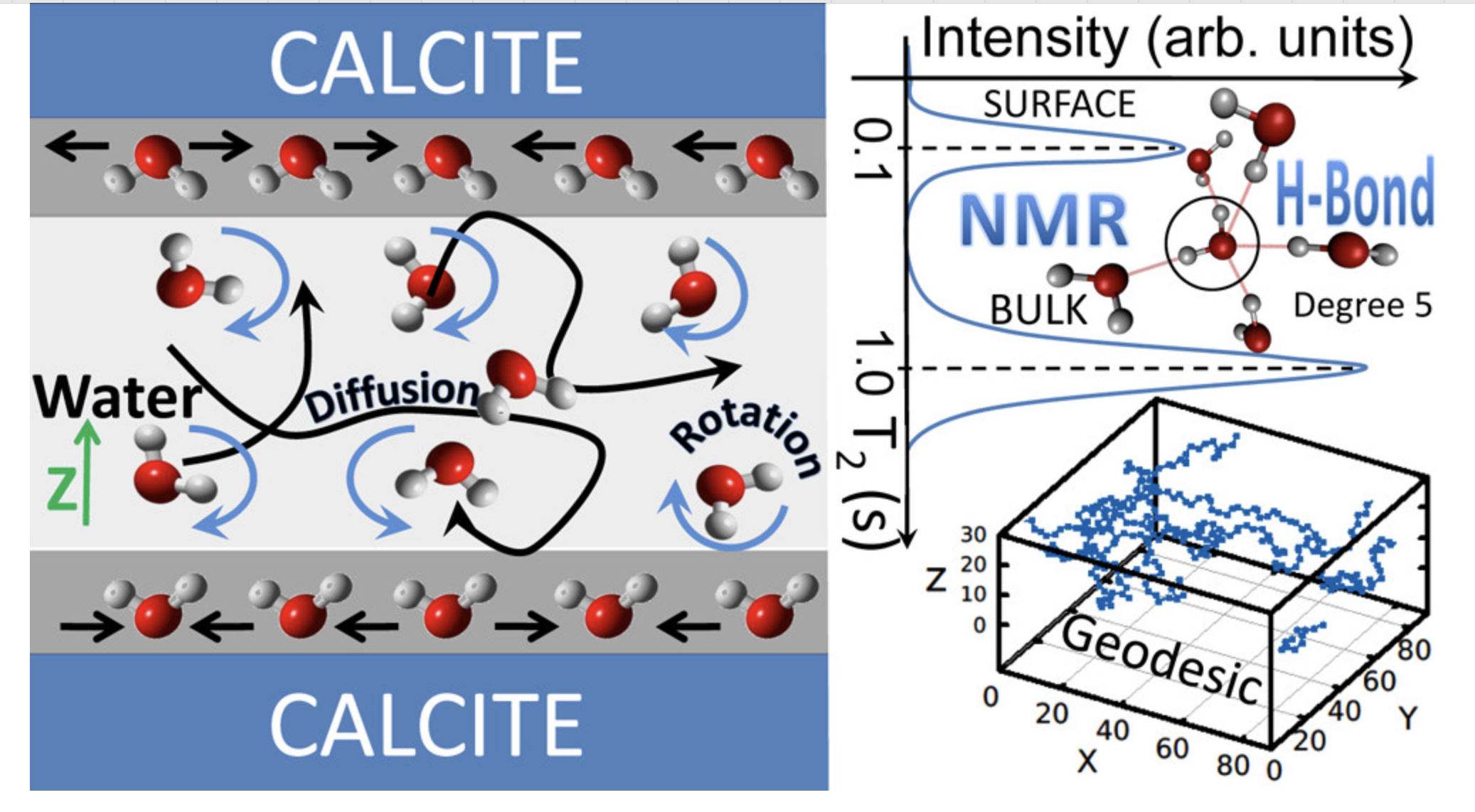
成键分析
分析分子动力学过程中的成键和断键事件,研究化学反应动力学。
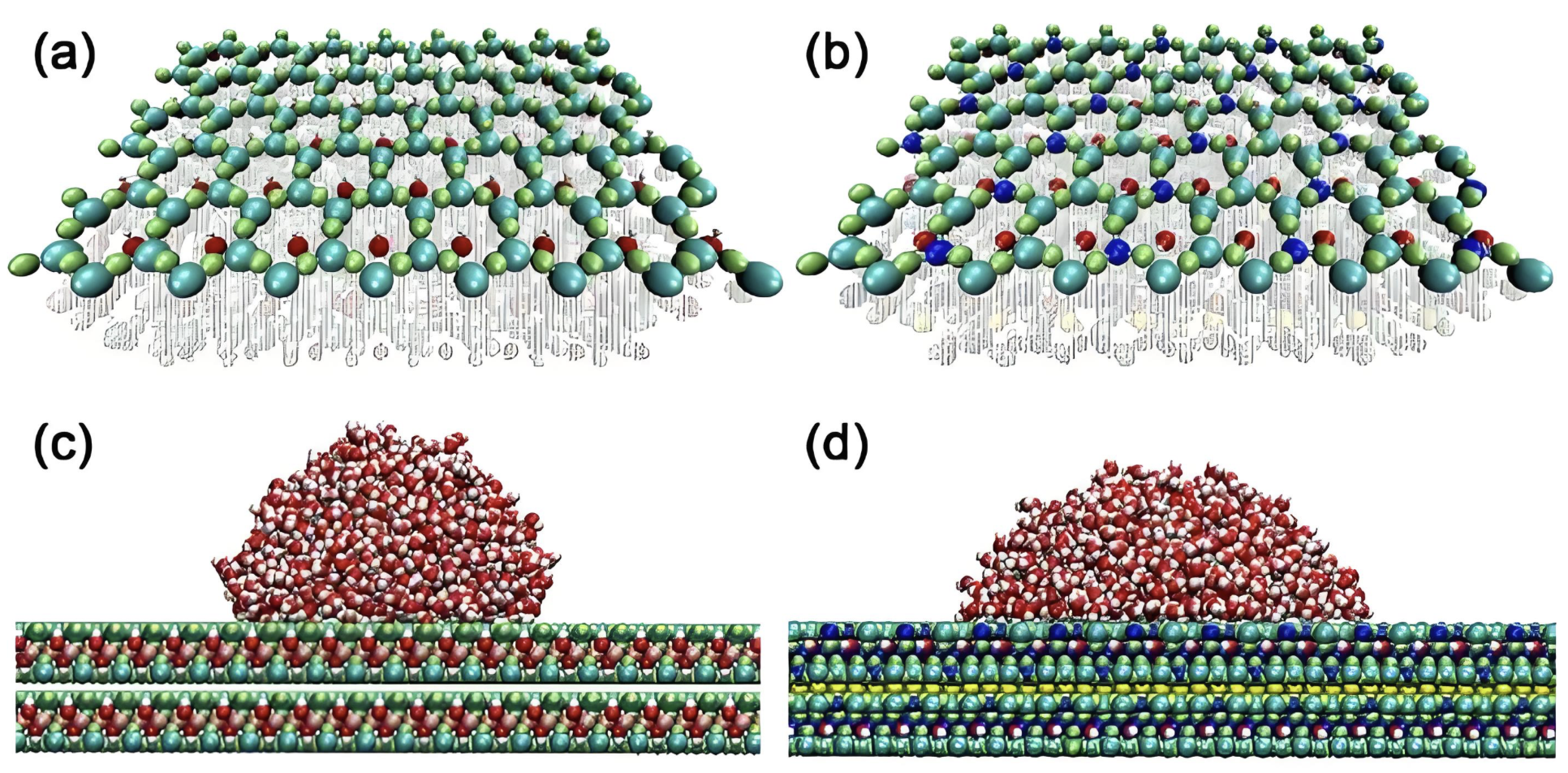
界面润湿
研究液体在固体表面的润湿行为,计算接触角和表面张力。
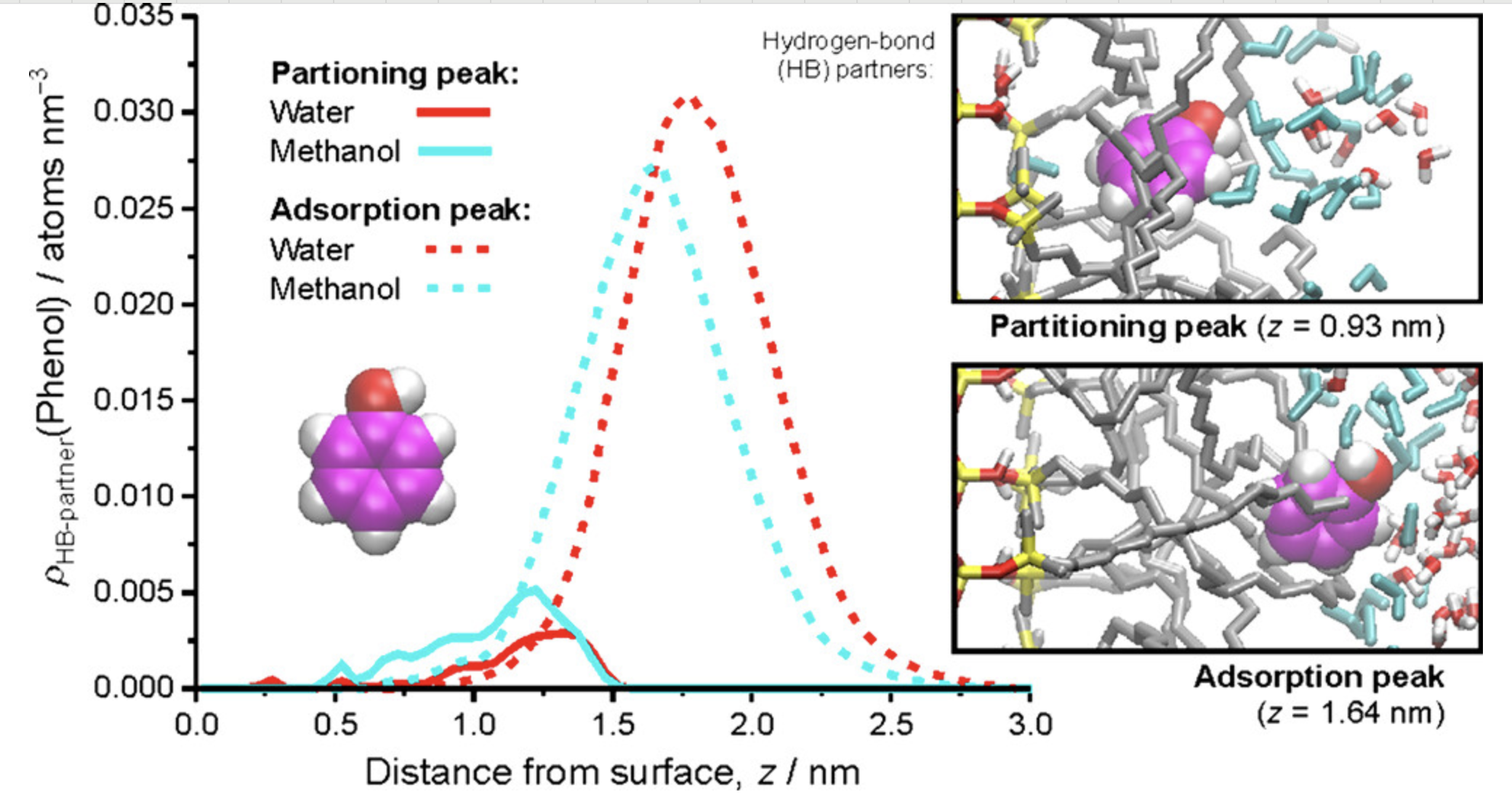
密度分布
分析体系中原子或分子的密度空间分布,研究相分离和界面结构。
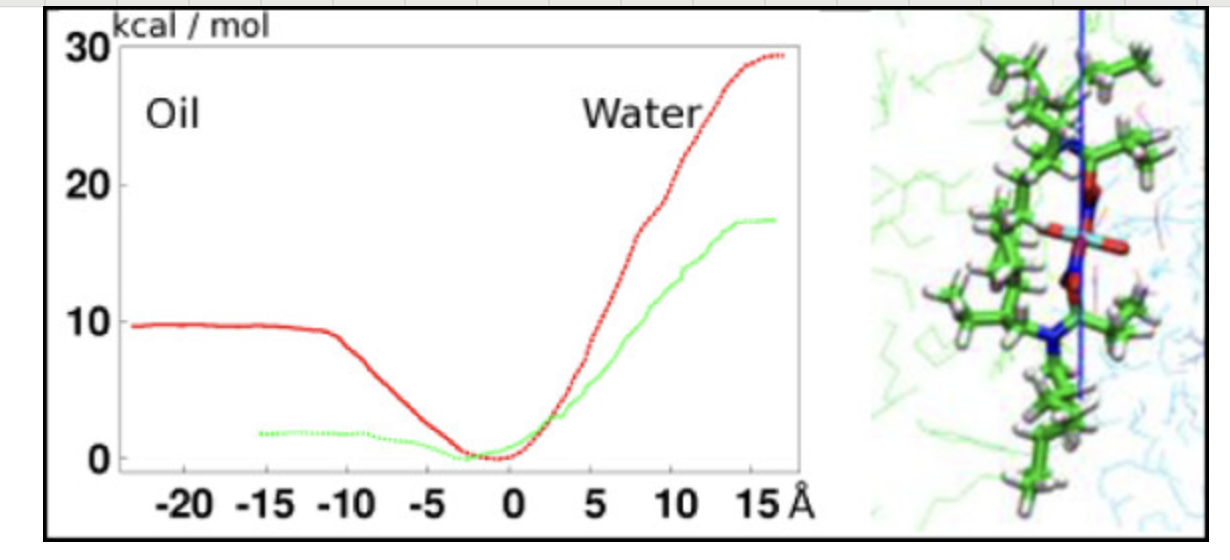
平均力势
计算分子间或原子间的平均力势能面,理解相互作用机理。
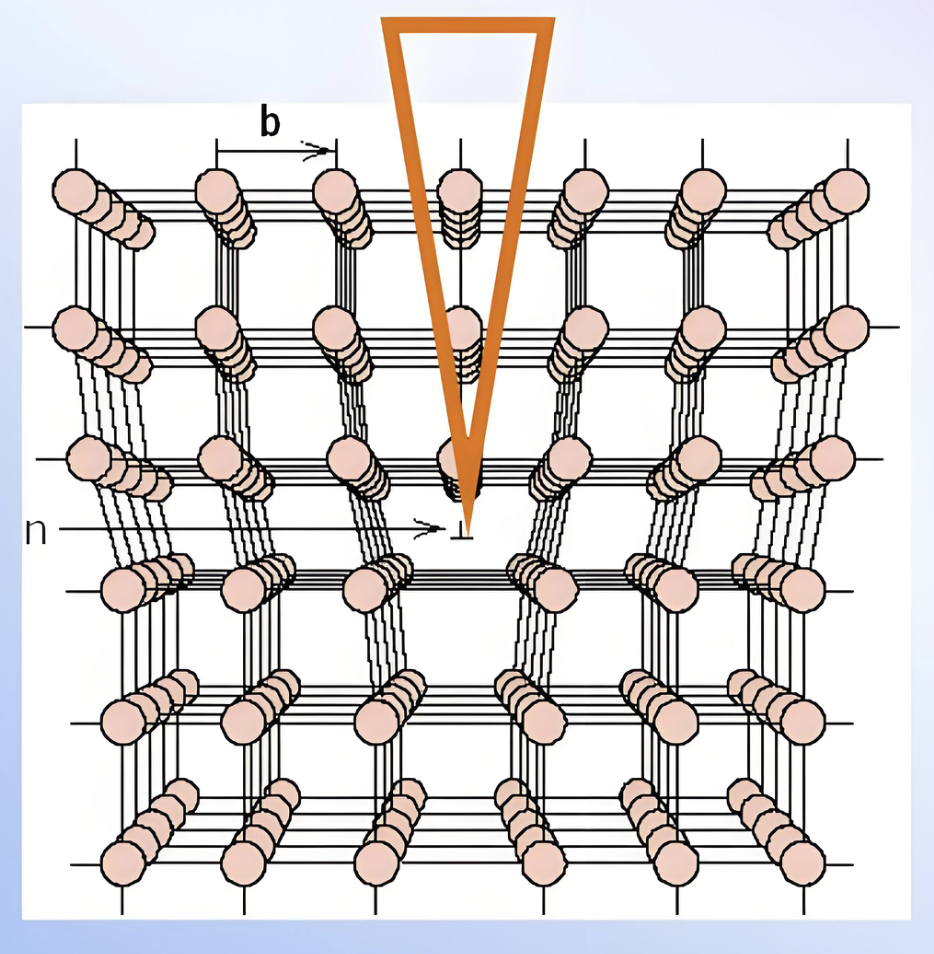
晶格位错
模拟晶体中位错的产生、运动和相互作用,研究塑性变形机理。
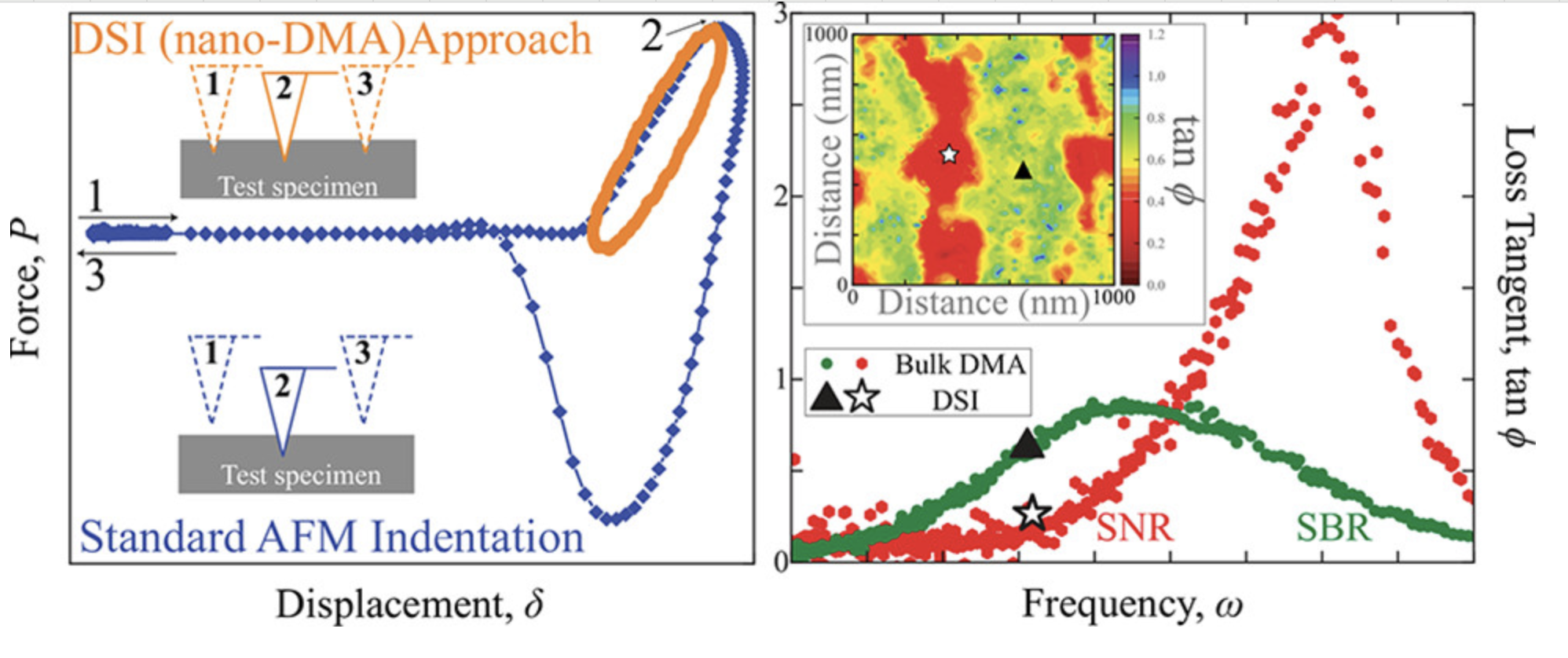
压痕/切削
模拟材料的压痕和切削过程,研究表面硬度和加工性能。
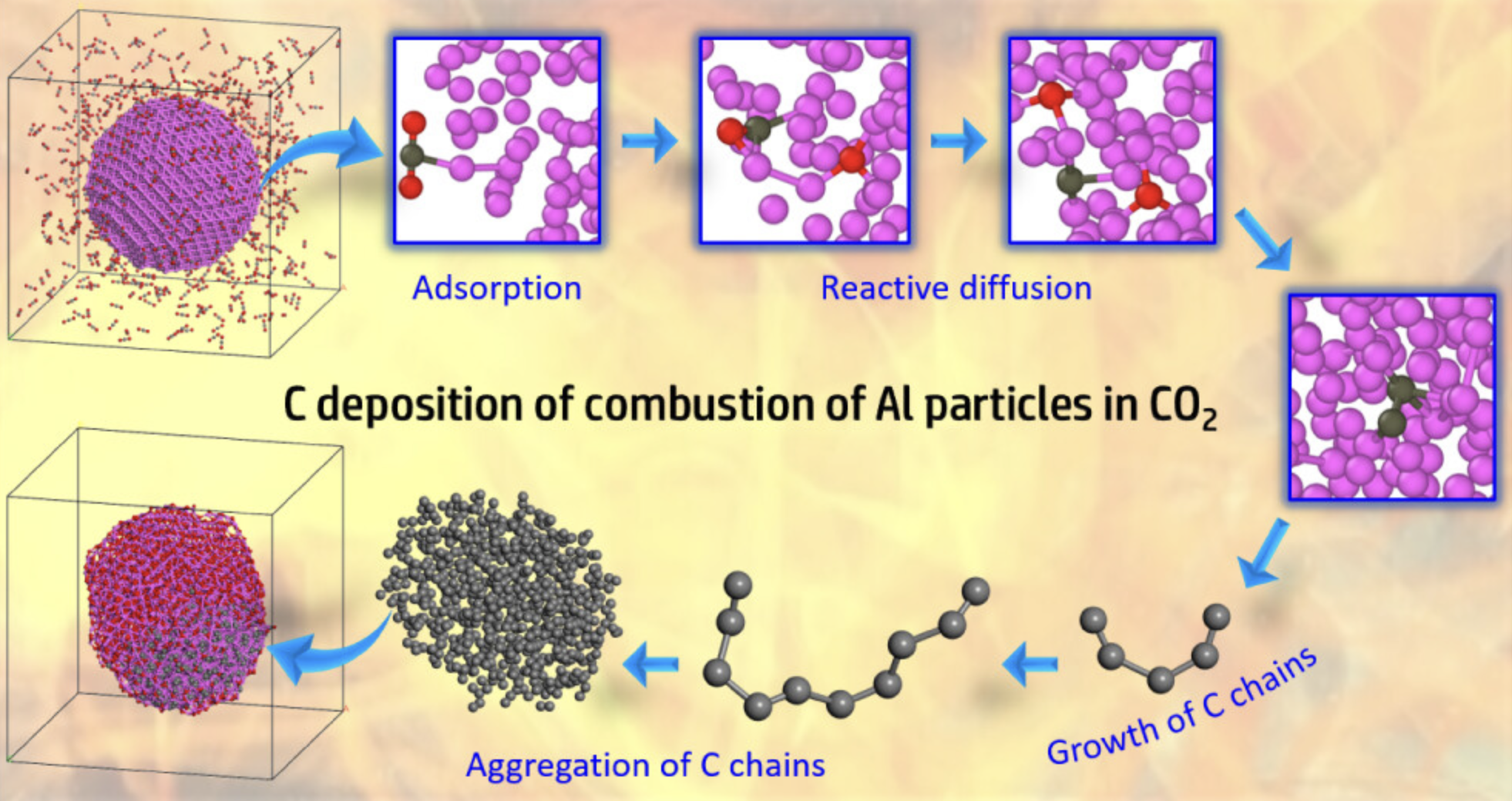
燃烧/热解
模拟有机材料的燃烧和热解过程,研究反应机理和产物分布。
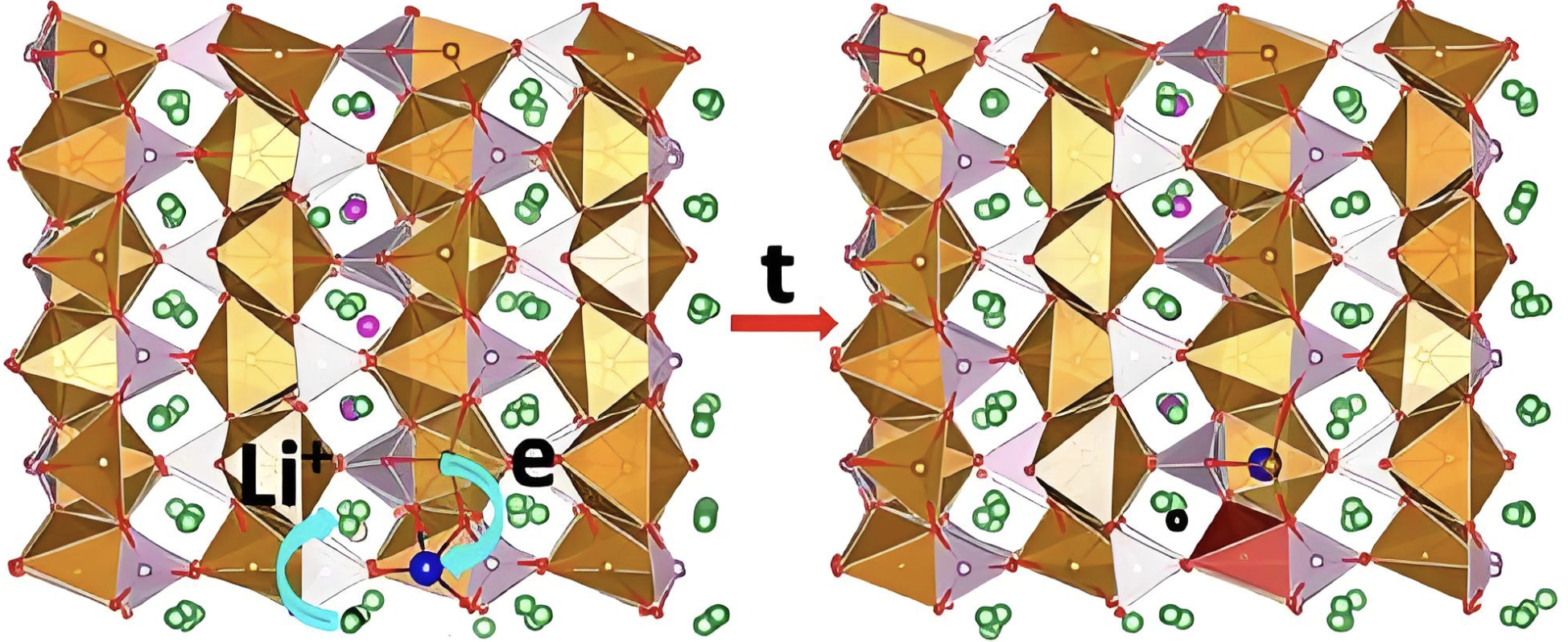
离子扩散
研究离子在固体电解质中的扩散路径和扩散机理。
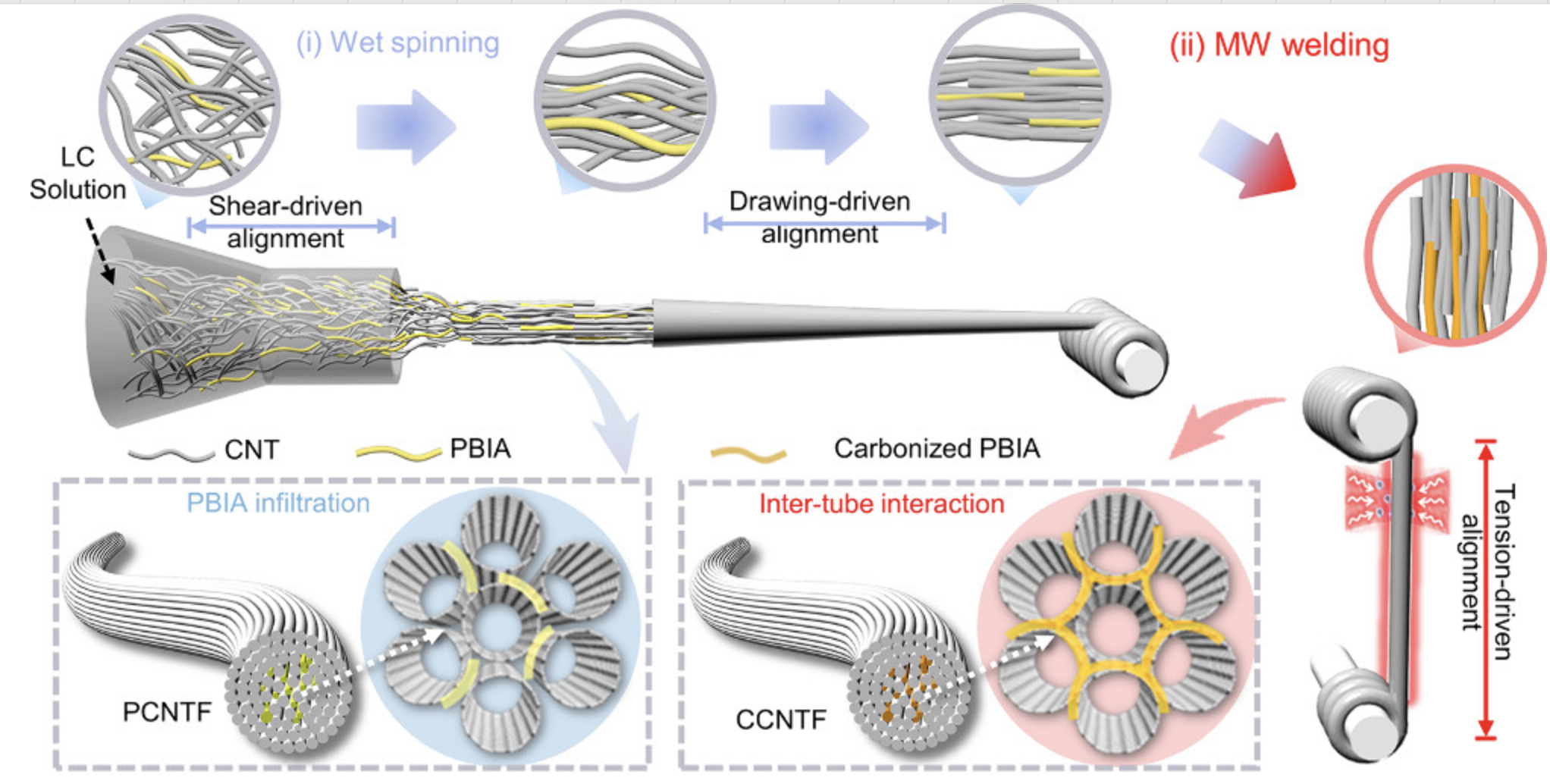
焊接
模拟金属焊接过程中的原子行为,研究焊缝组织和性能。
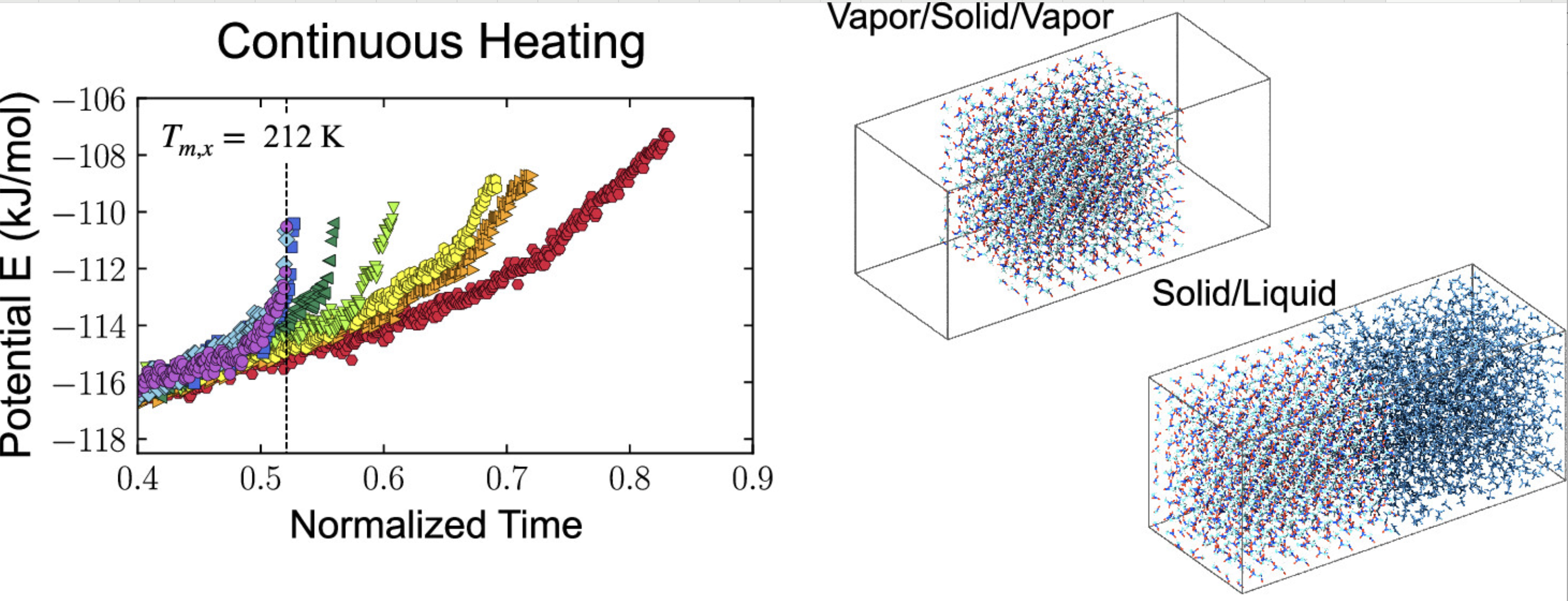
熔点
计算材料的熔点温度,通过固液两相共存模拟确定相变点。
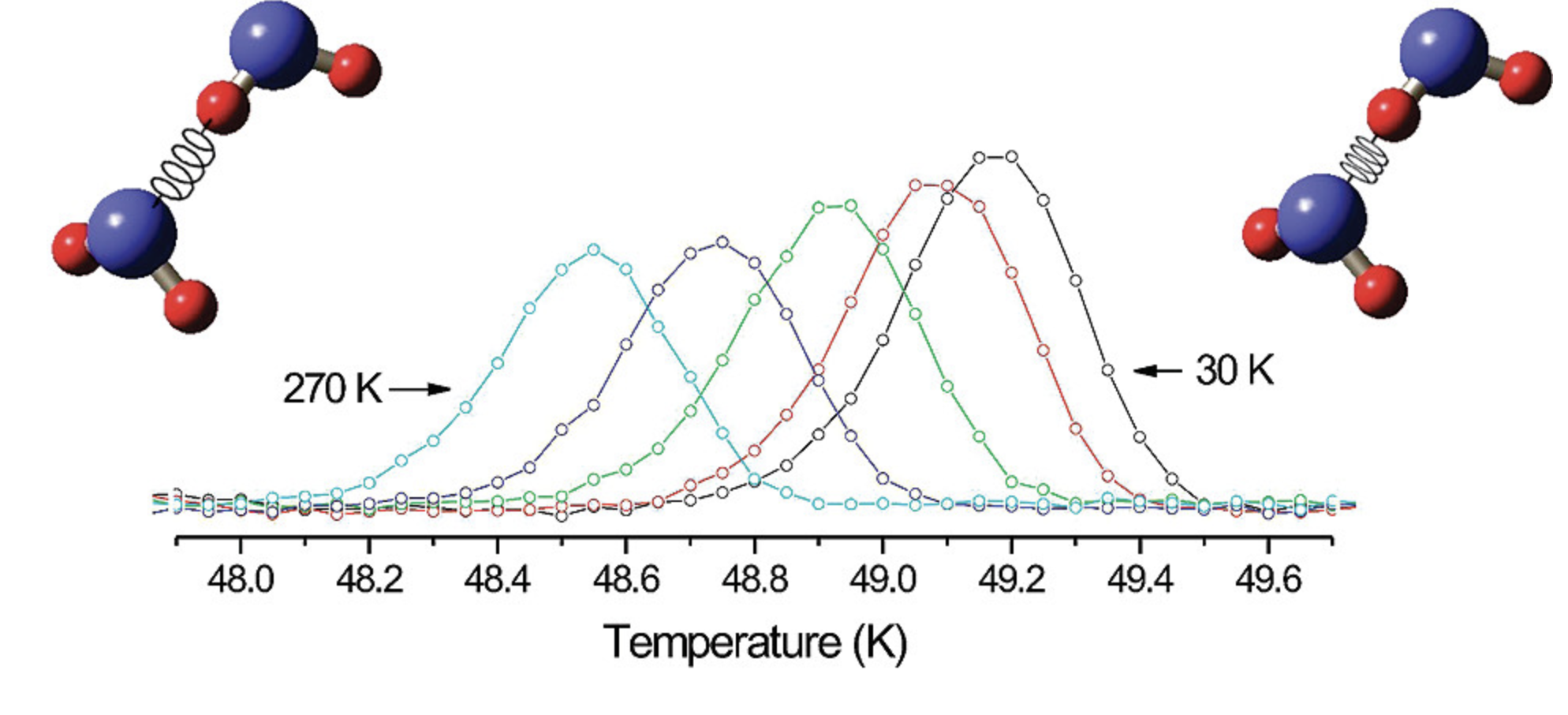
热膨胀率
计算材料的线性和体积热膨胀系数,评估热稳定性。
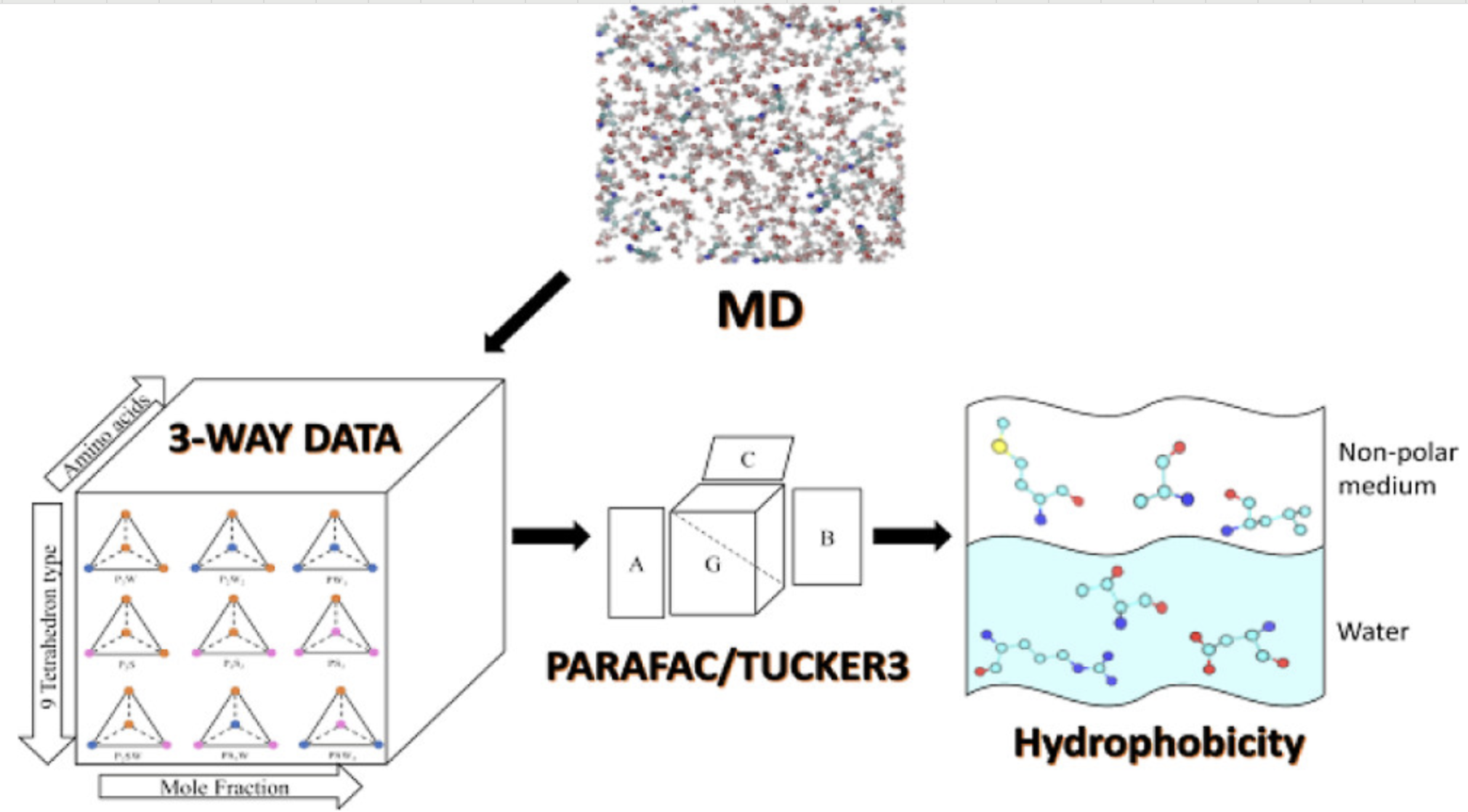
亲疏水性分析
分析表面的亲水疏水性质,通过水滴接触角等参数表征。
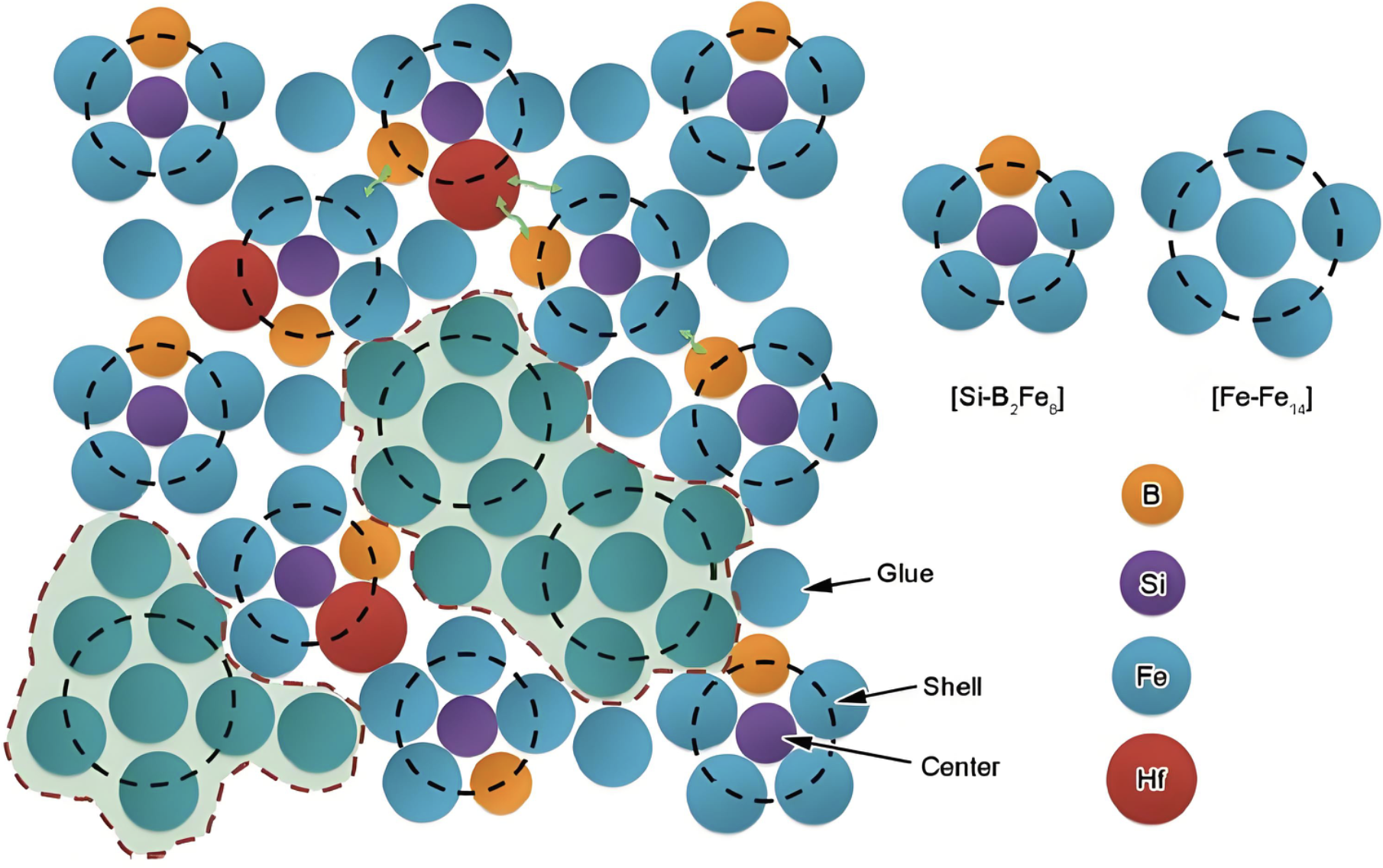
非晶结构
研究非晶态材料的短程和中程有序结构特征。
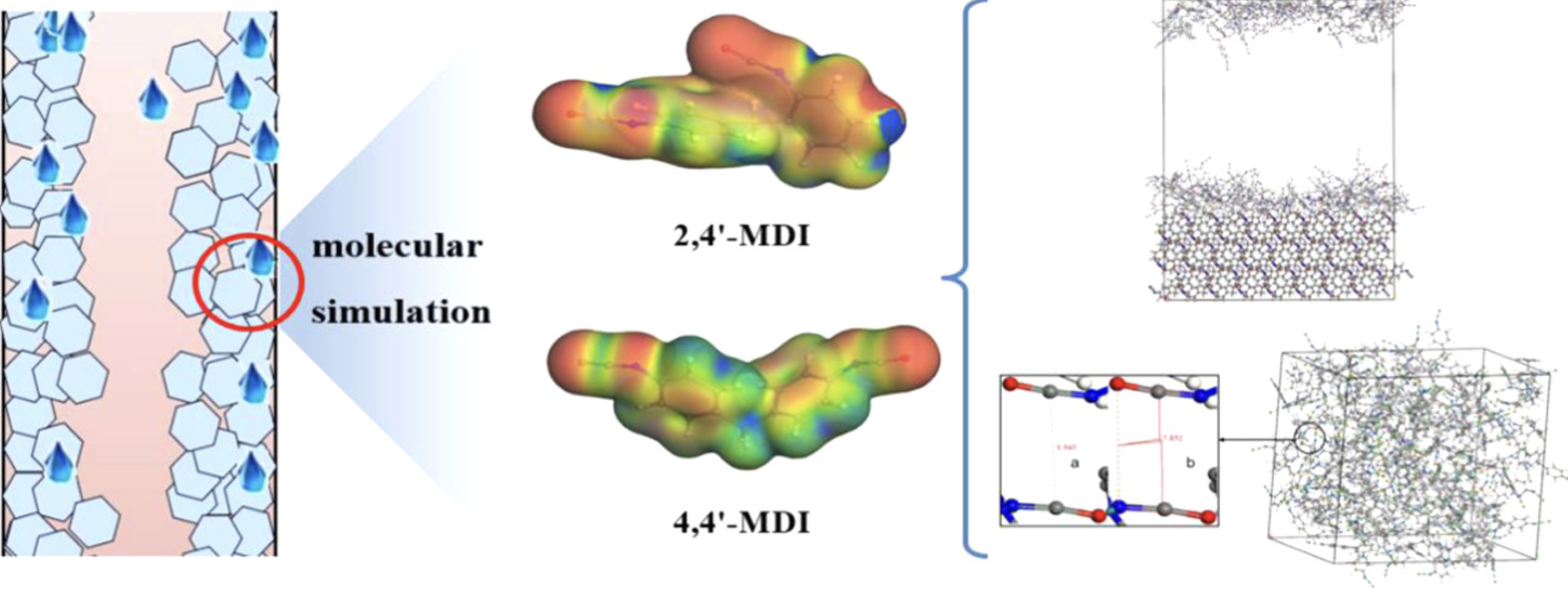
结晶
模拟材料的结晶过程,研究晶核形成和晶体生长动力学。
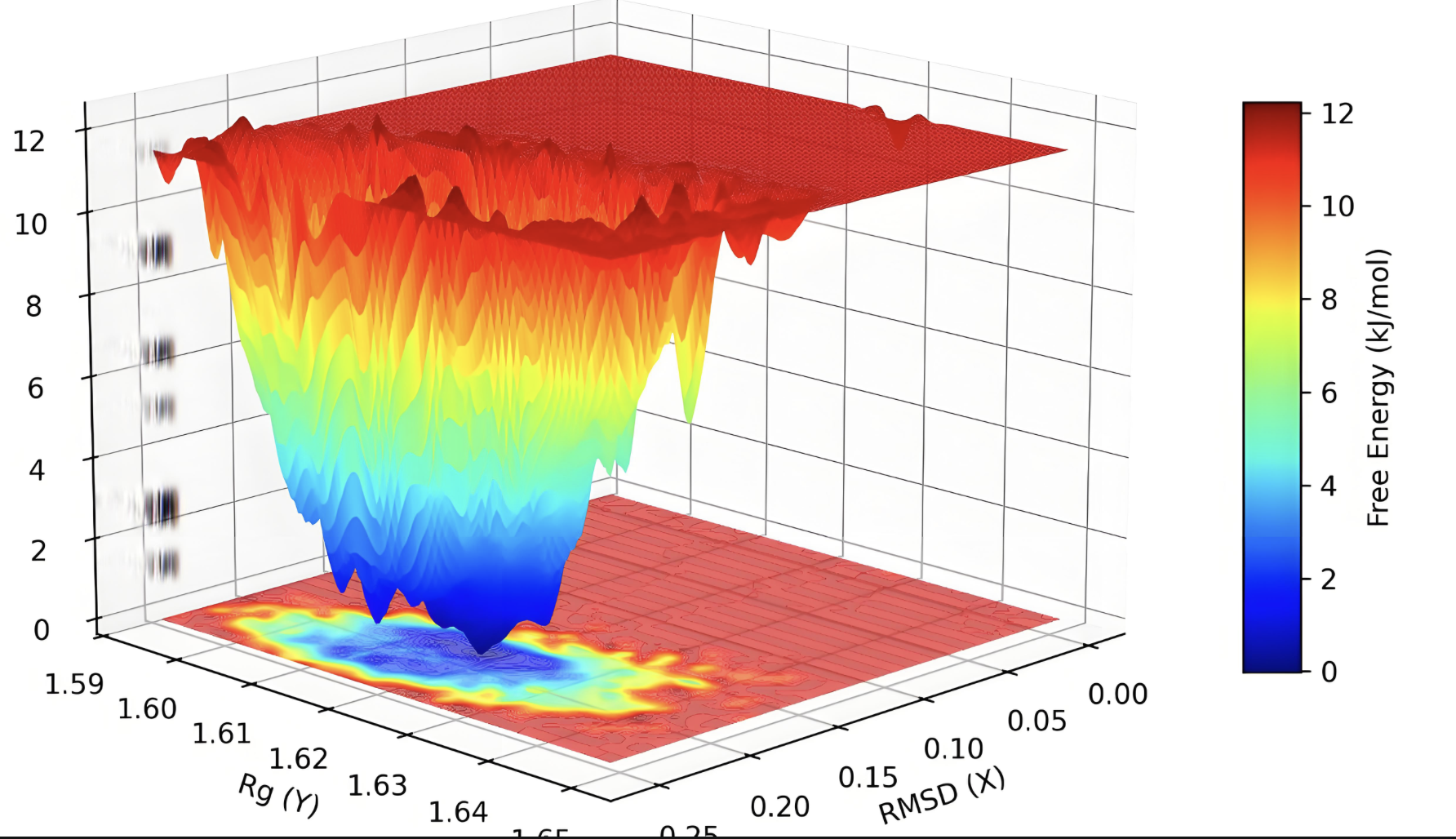
自由能形貌图
构建反应坐标的自由能面,分析反应路径和活化屏障。
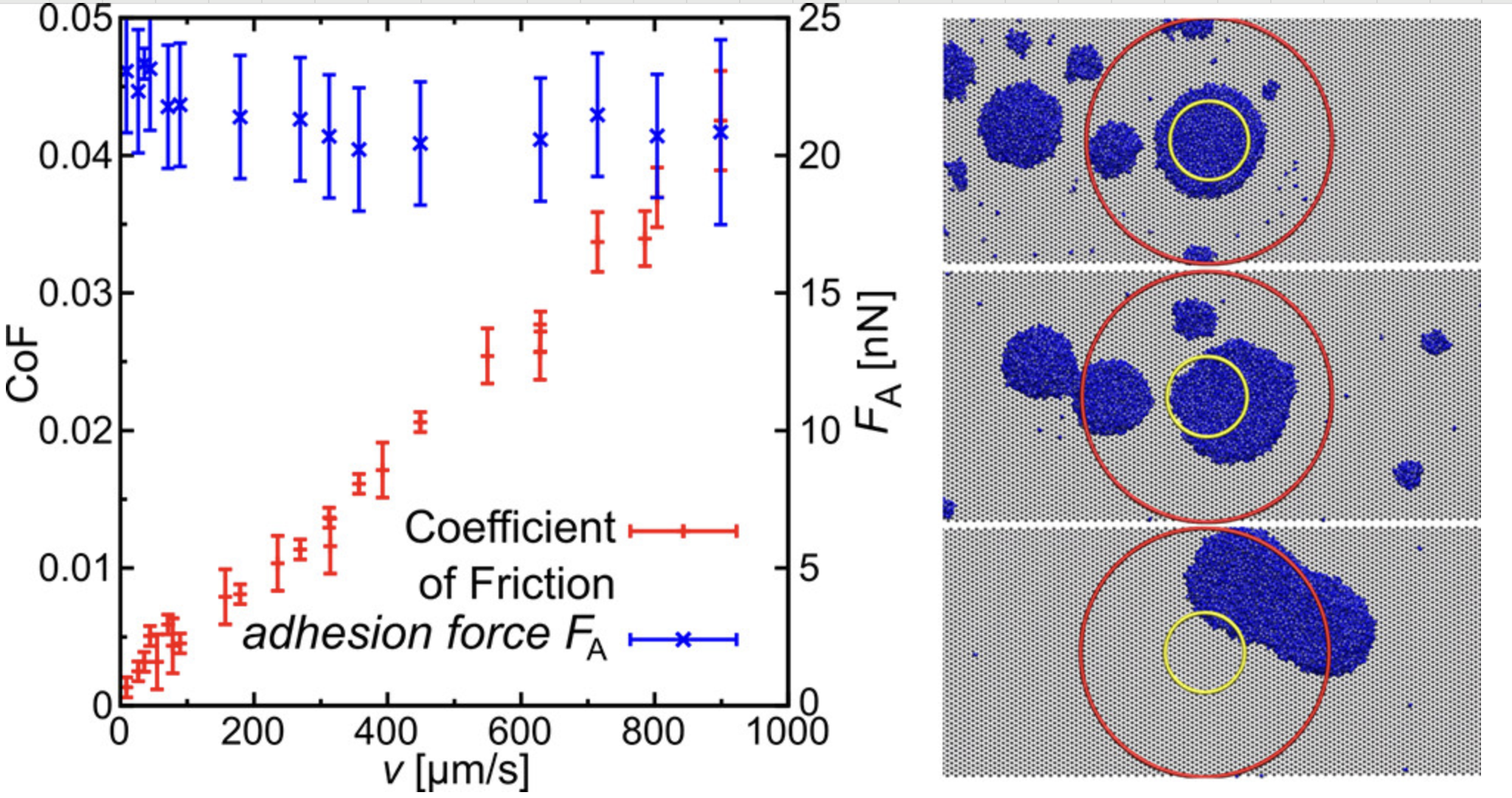
摩擦
模拟材料表面的摩擦行为,研究摩擦系数和磨损机理。
生物分子模拟
针对生物大分子系统的专业计算,包括蛋白质、核酸、膜结构等复杂生物体系
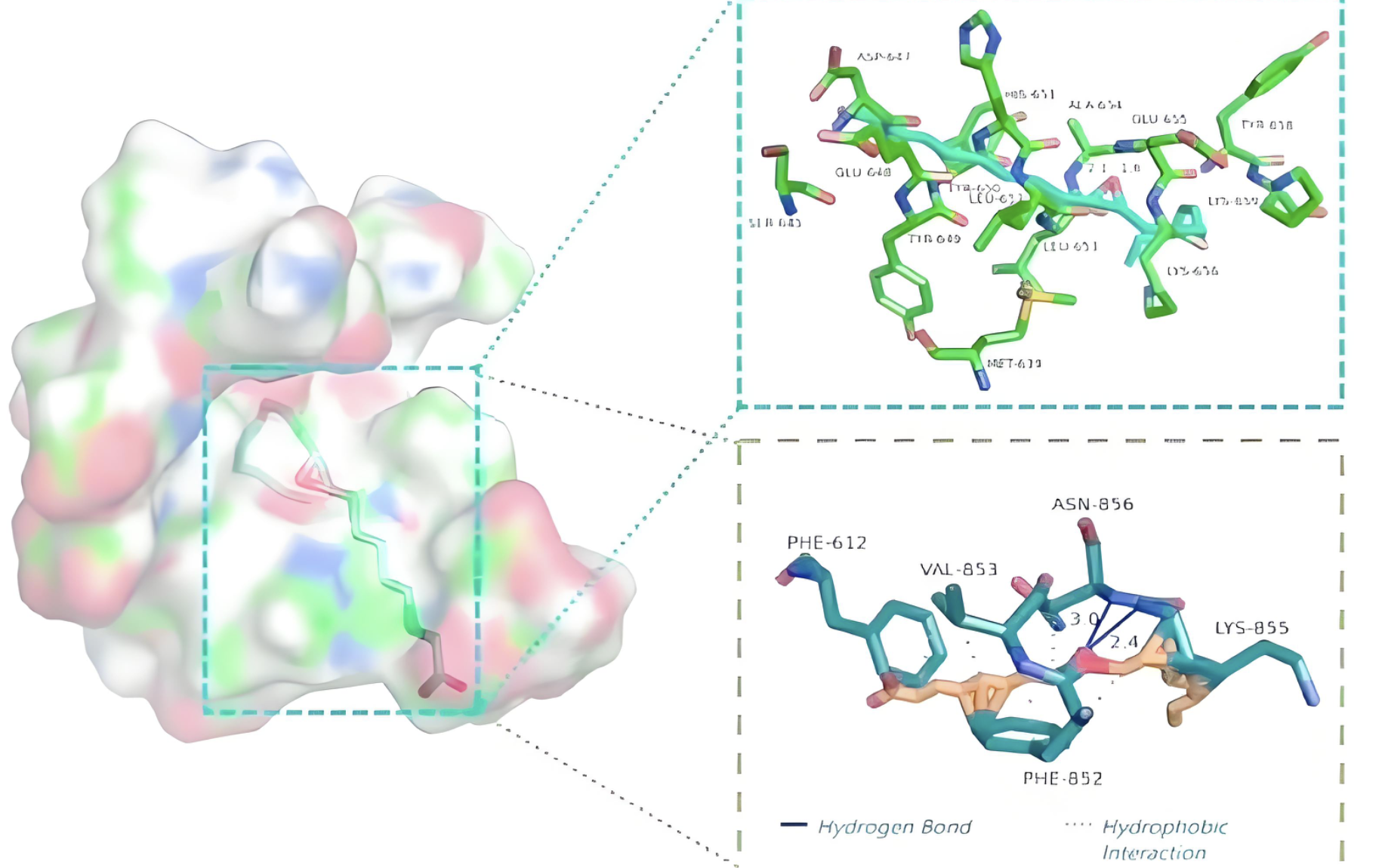
分子对接
预测小分子与蛋白质的结合模式,用于药物设计和酶活性研究。
虚拟筛选
通过计算方法快速筛选潜在药物分子,降低实验成本。
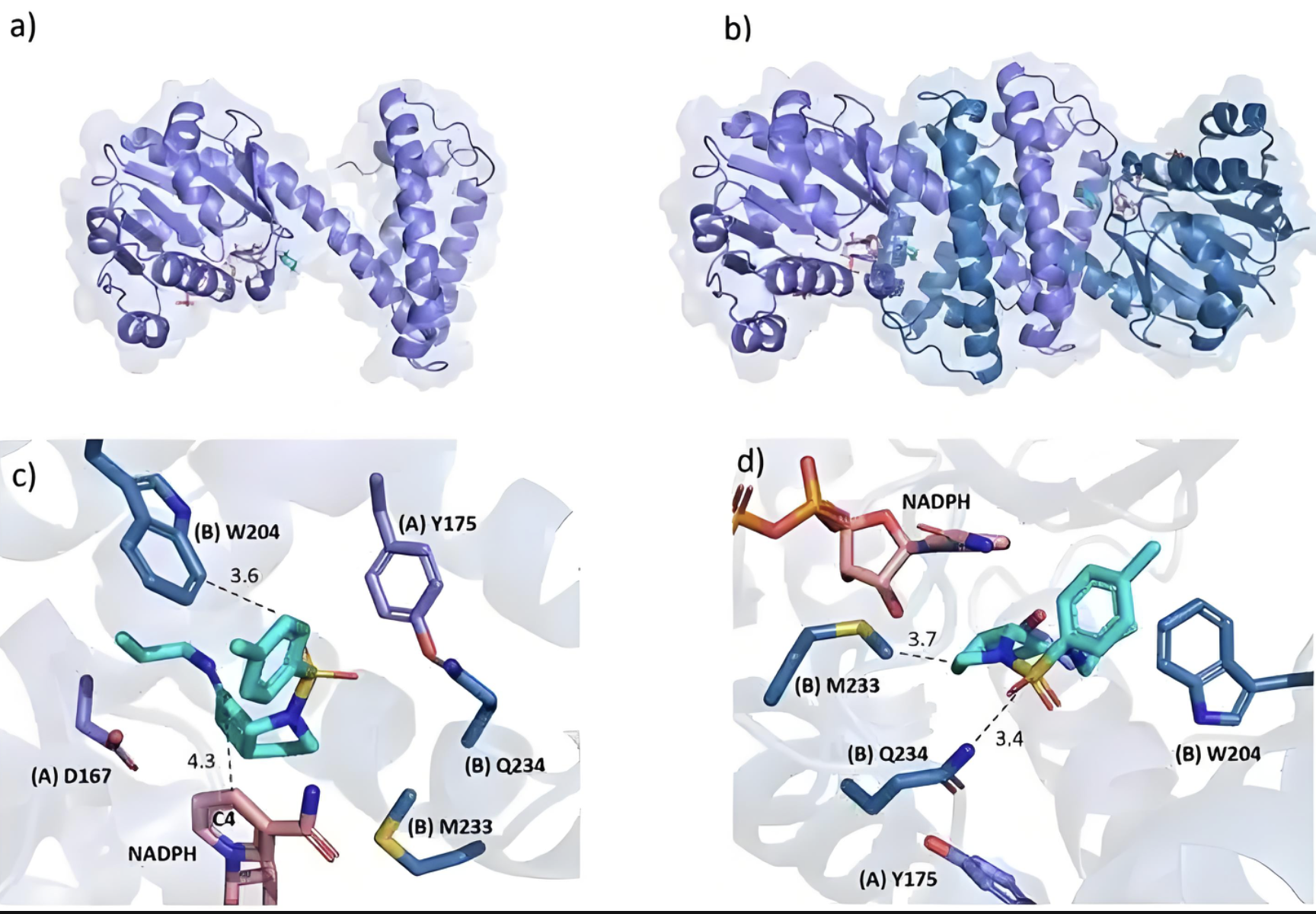
药物设计
基于结构的药物设计,优化小分子化合物的活性和选择性。
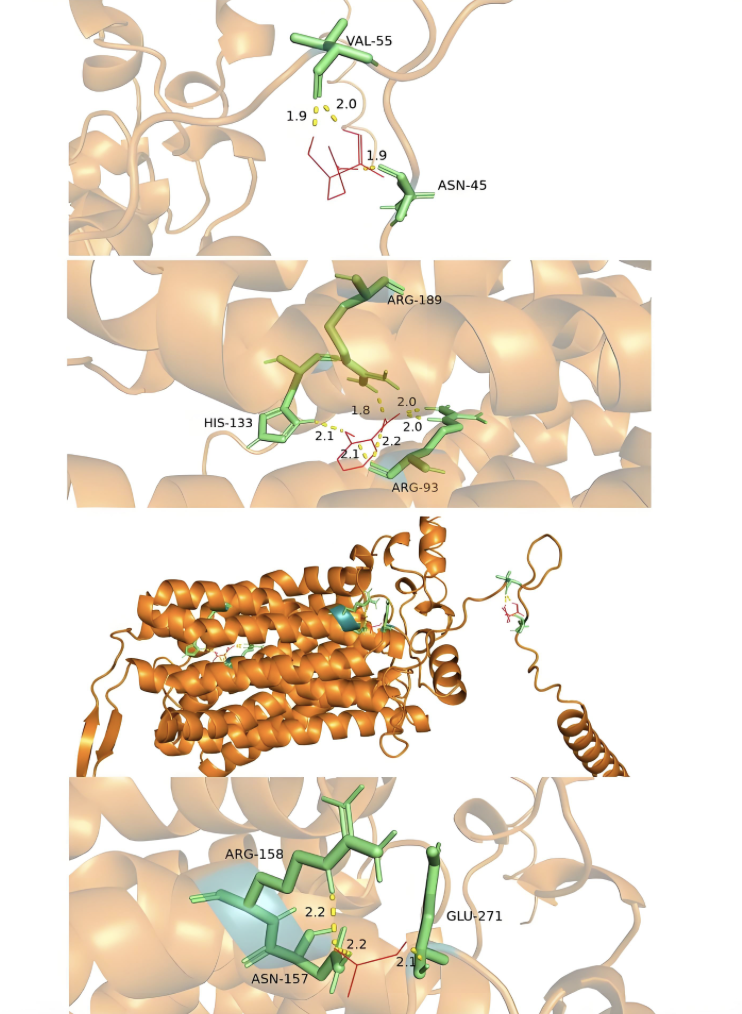
分子间相互作用
分析生物分子间的氢键、静电相互作用等弱相互作用力。
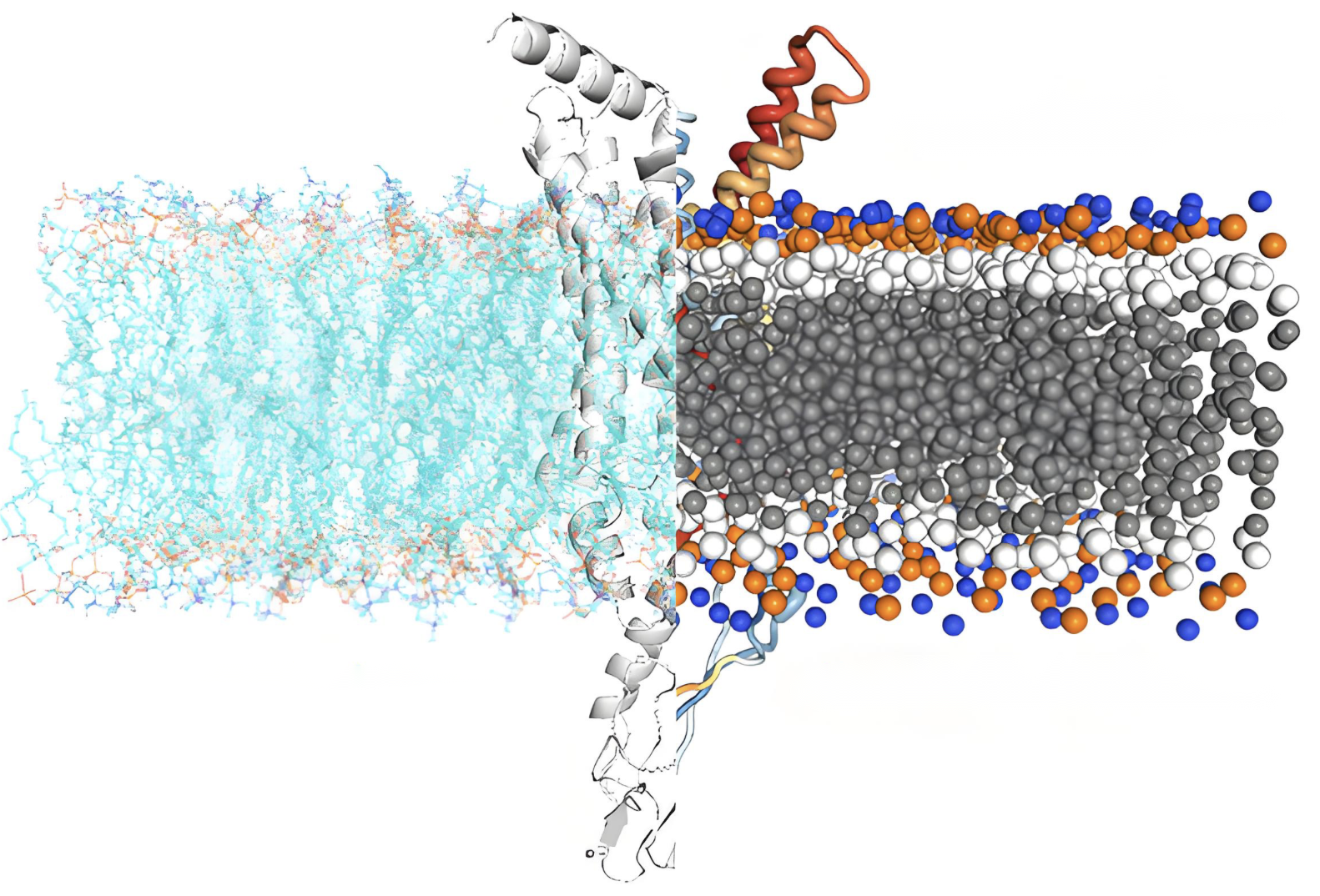
粗粒化模拟
采用粗粒化模型研究大尺度生物系统的动力学行为。
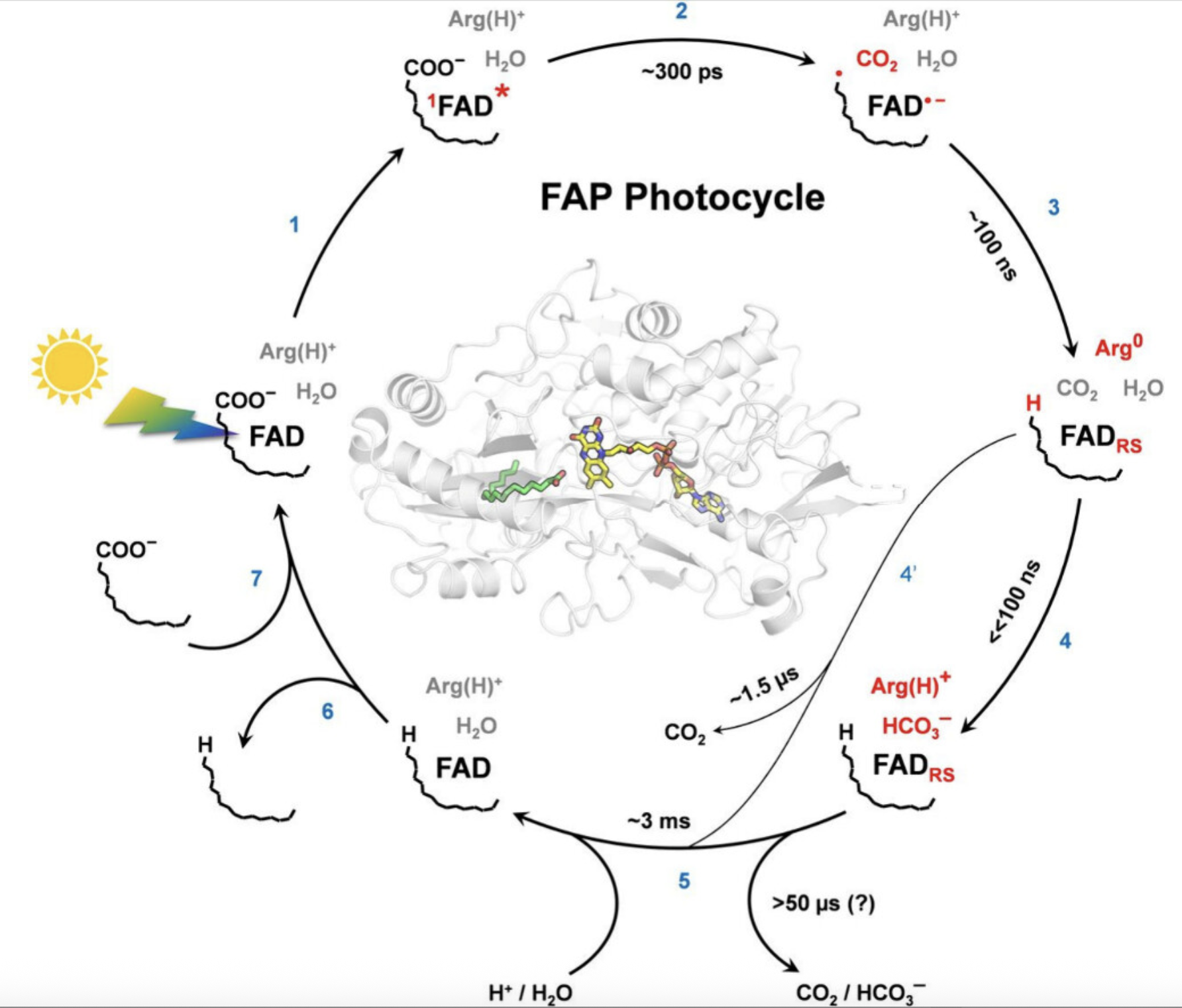
酶催化
研究酶催化反应的机理,分析活性位点和催化效率。
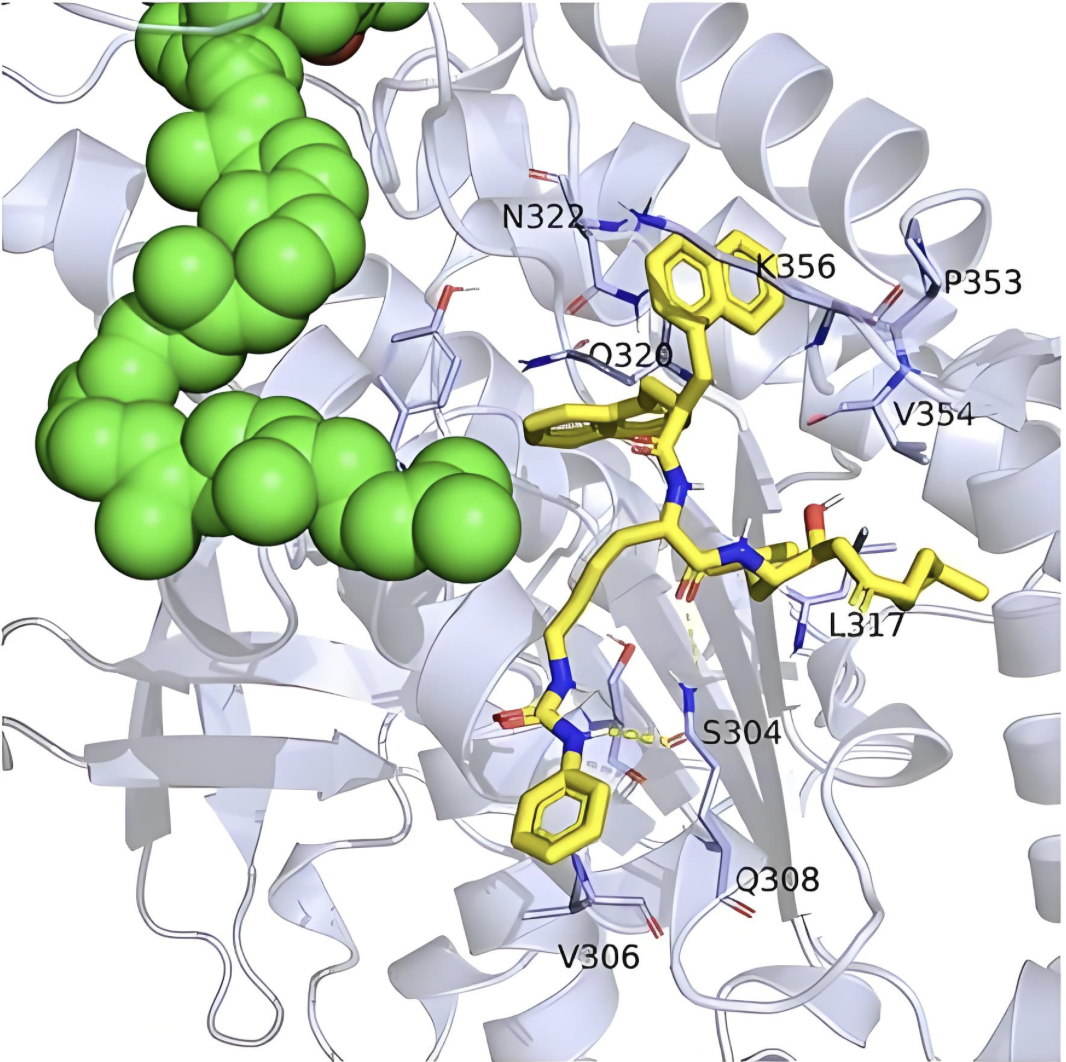
生物蛋白分子动力学
模拟蛋白质在生理条件下的动力学行为和构象变化。
QM/MM
结合量子力学和分子力学方法,研究生物系统中的化学反应。
同源建模
基于已知结构预测蛋白质三维结构,用于结构功能研究。
配体-受体结合
研究配体与蛋白质的结合过程和结合强度。
病毒研究
研究病毒蛋白结构和病毒-宿主相互作用机制。
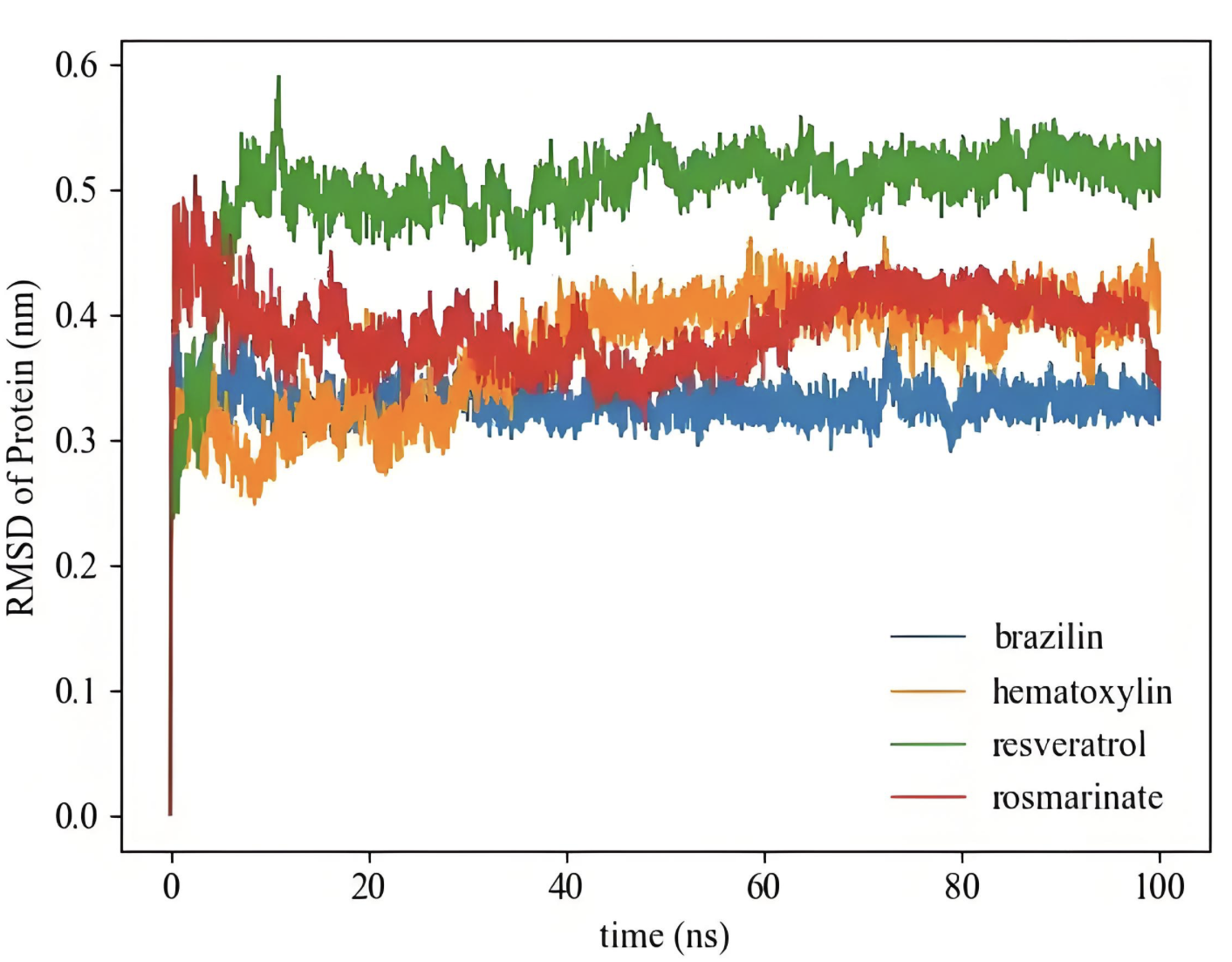
RMSD
计算结构的均方根偏差,评估结构稳定性和构象变化。
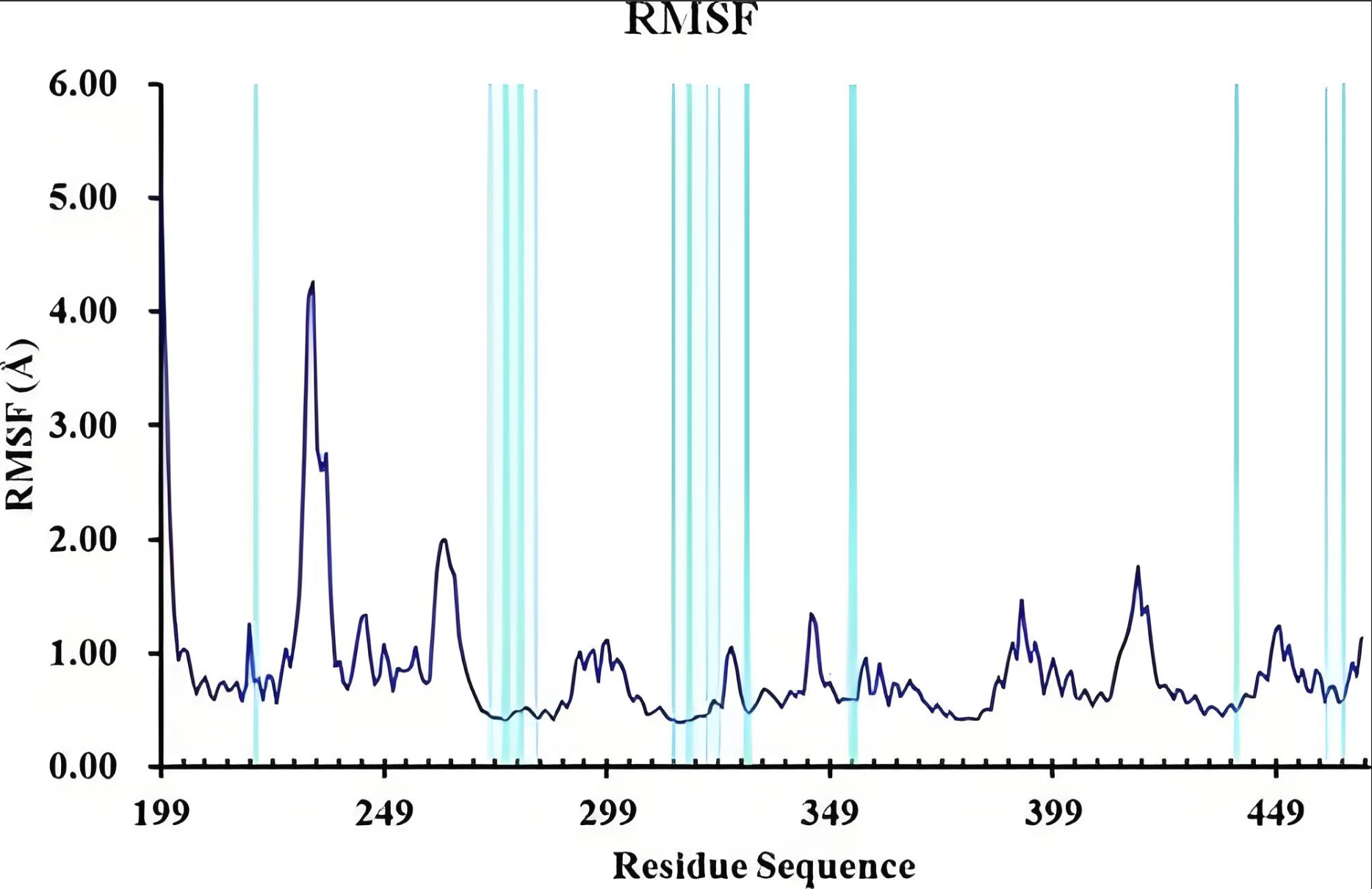
RMSF
计算原子位置的均方根涨落,分析结构柔性和动态性。
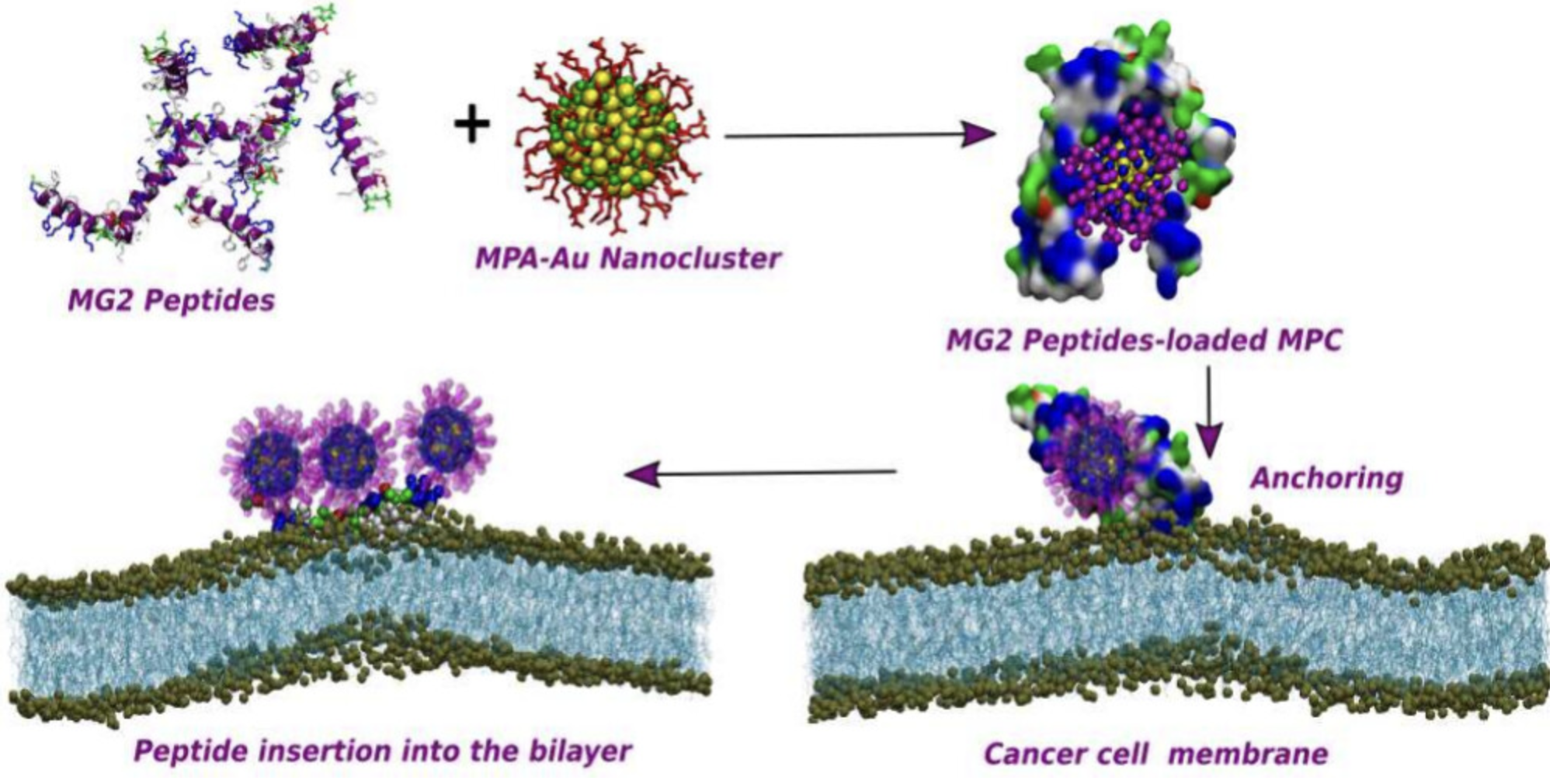
多肽设计
设计具有特定功能的多肽序列,优化结构和活性。
生信分析
生物信息学数据分析,包括序列比对、系统发育分析等。
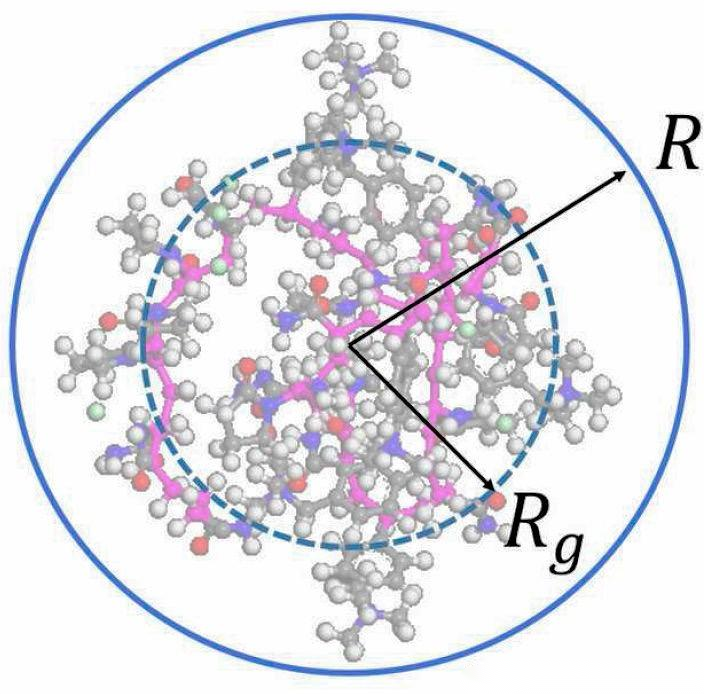
回旋半径
计算蛋白质的回旋半径,评估结构紧密程度和折叠状态。
结合自由能
计算配体与蛋白质的结合自由能,预测结合亲和力。
细胞膜转运
研究分子通过细胞膜的转运机制和渗透性。
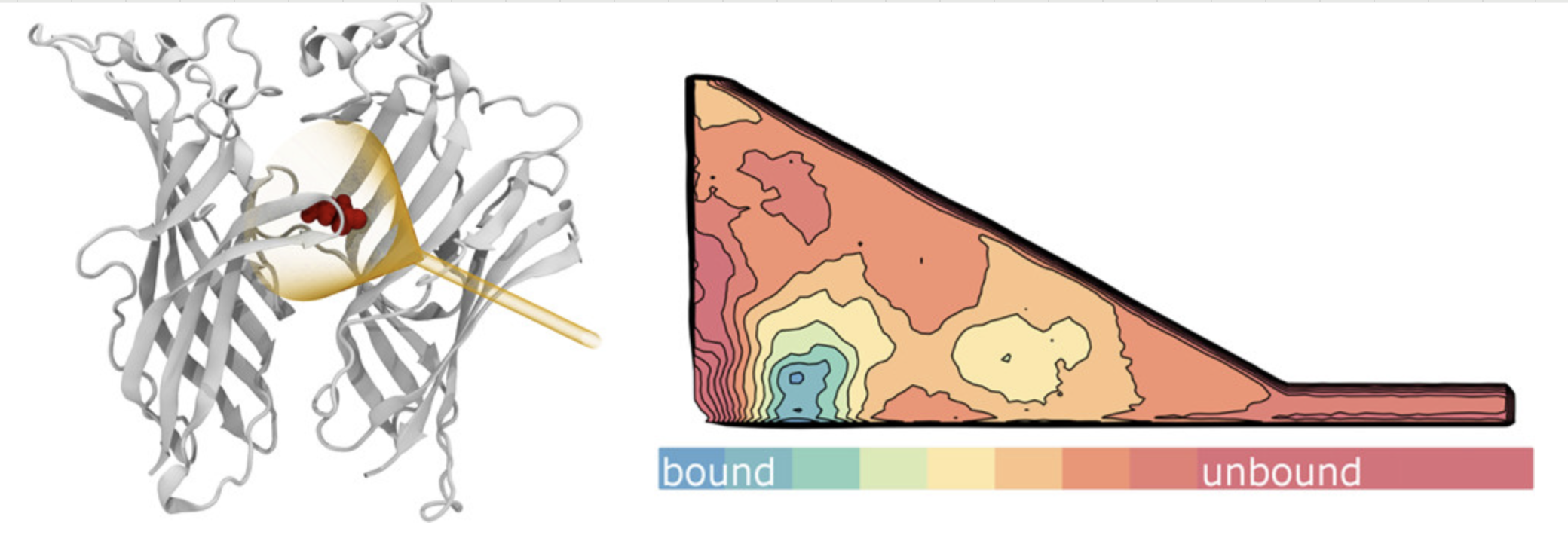
配体通道
分析配体进入和离开蛋白质活性位点的通道路径。
氢键分析
分析生物分子中氢键的数量、强度和动态变化。
蛋白质折叠
研究蛋白质三维结构的形成过程和稳定性因素。
QSAR模型预测
建立定量构效关系模型,预测分子的生物活性。
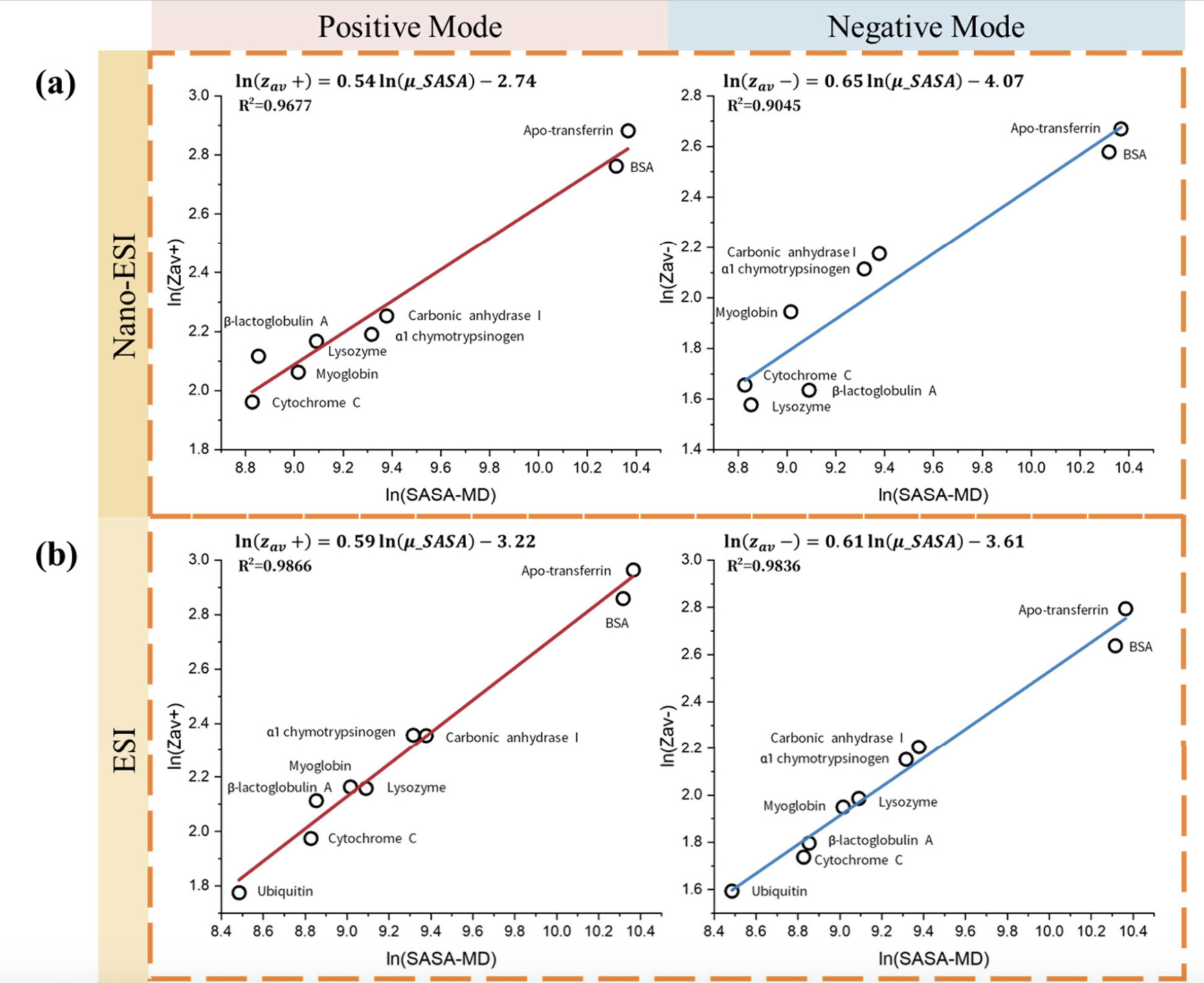
SASA分析
计算溶剂可及表面积,分析蛋白质表面暴露程度。
成药性分析
评估化合物的成药性质,包括Lipinski规则等药代动力学性质。
机器学习
结合人工智能技术,加速材料发现和性能预测,提供智能化的计算解决方案
高通量筛选
自动化大规模材料筛选,快速识别具有特定性能的候选材料。
机器学习势
基于机器学习训练的原子间势函数,提供DFT精度的分子动力学模拟。
神经网络势
使用神经网络构建高精度势函数,实现大体系的量子精度模拟。
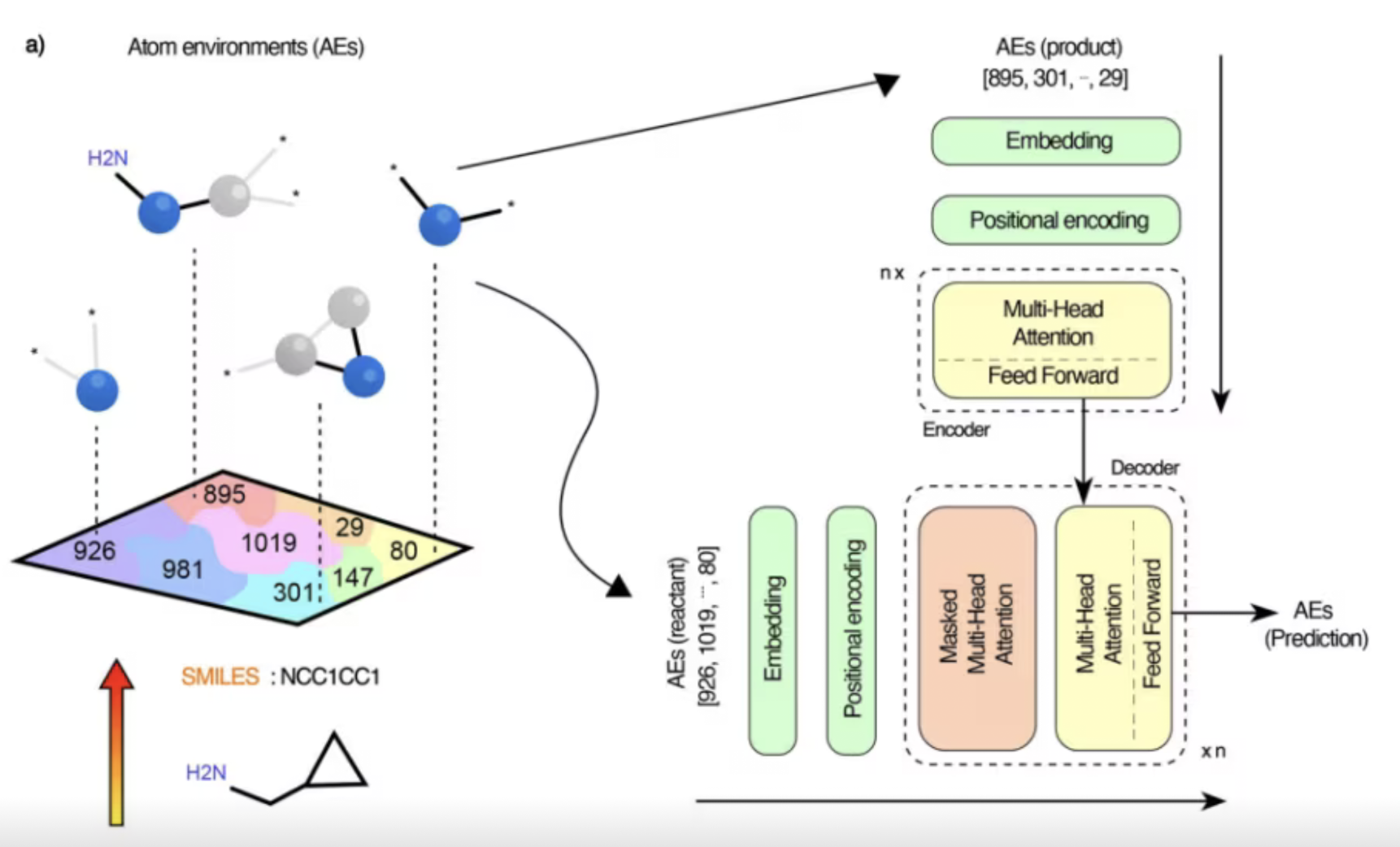
预测反应路径
使用机器学习预测化学反应的最优路径和过渡态结构。
半导体结构筛选
针对半导体材料的成分优化和性能预测。
数据分析与挖掘
从大量计算数据中提取有价值的信息和规律。
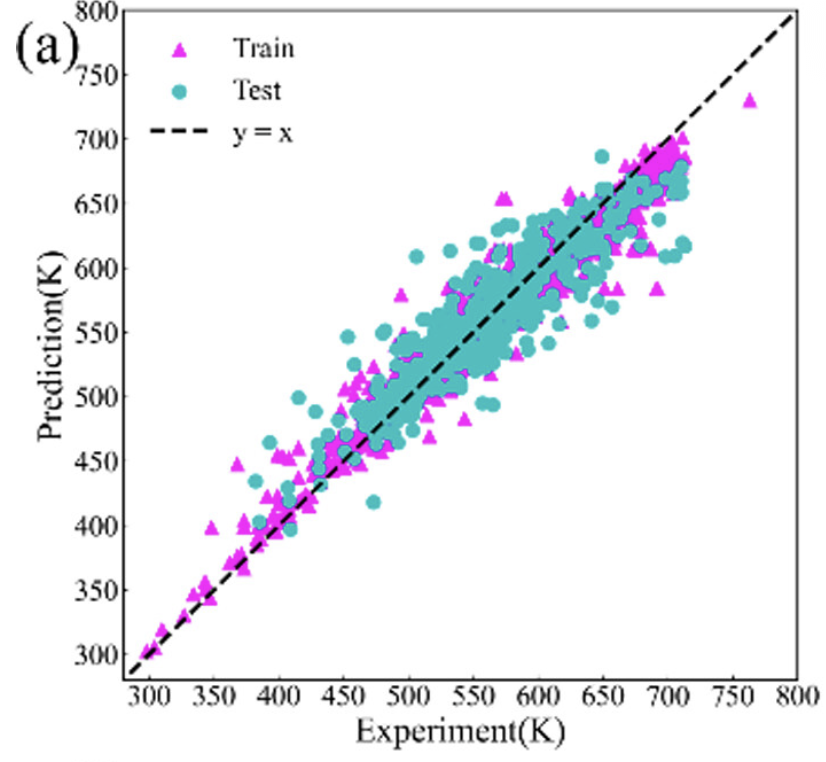
随机森林
使用随机森林算法进行材料性质预测和分类。
预测催化性能
基于描述符和机器学习模型预测催化剂的活性和选择性。
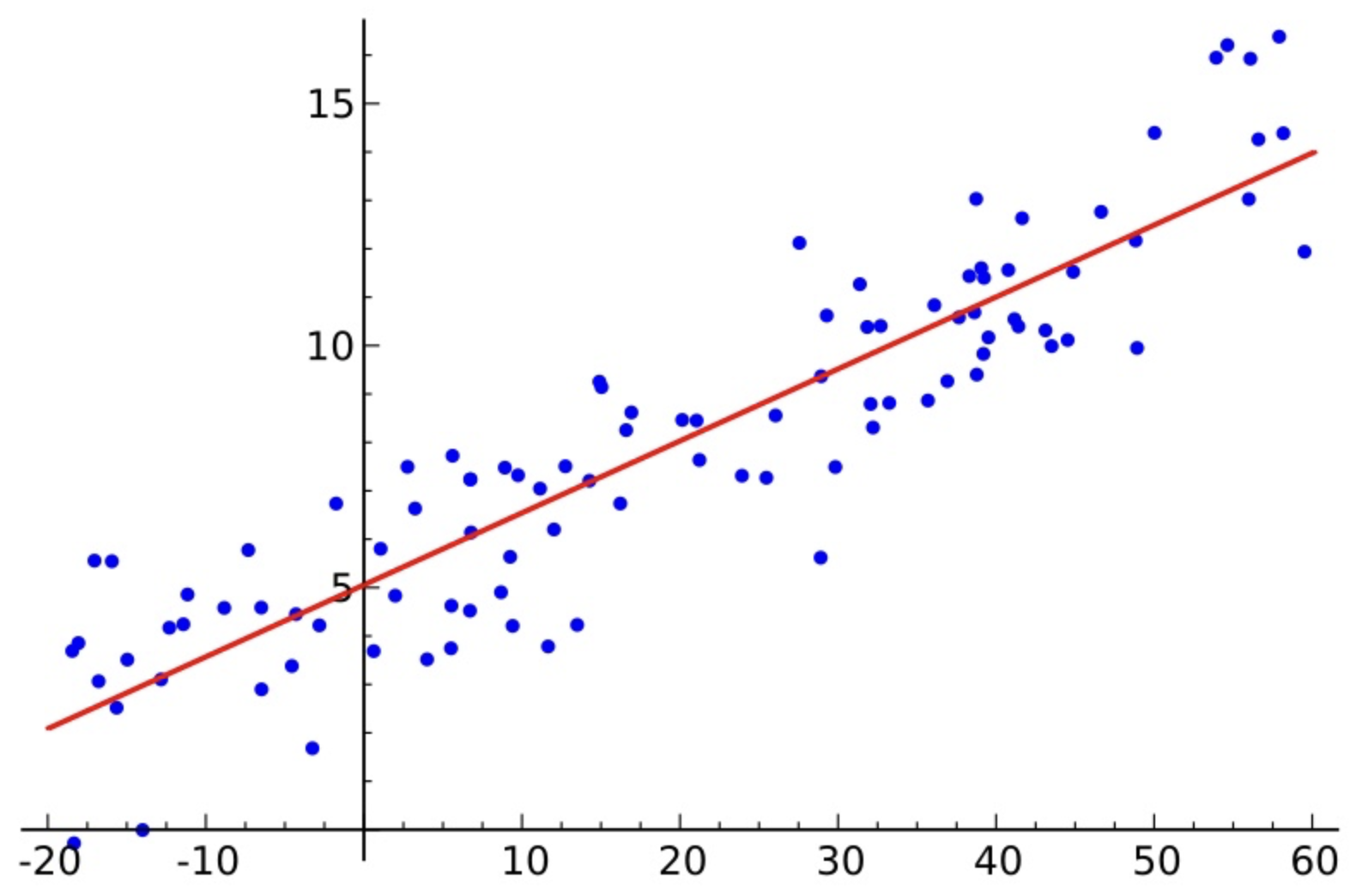
线性回归
使用线性回归模型建立材料性质与结构参数的关系。
预测结构稳定性
使用机器学习预测材料结构的热力学和动力学稳定性。
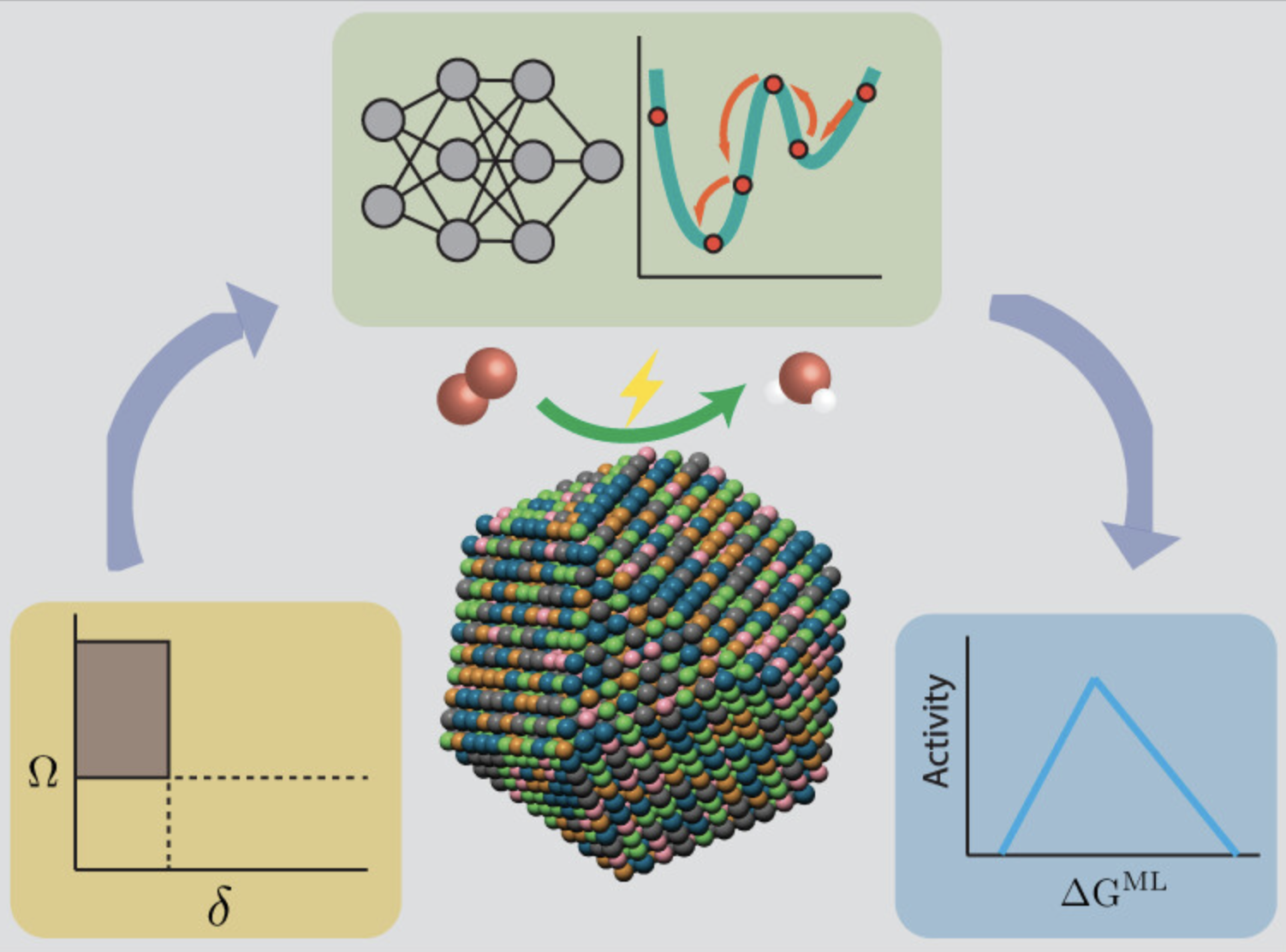
高熵合金材料筛选
针对高熵合金的成分优化和性能预测。
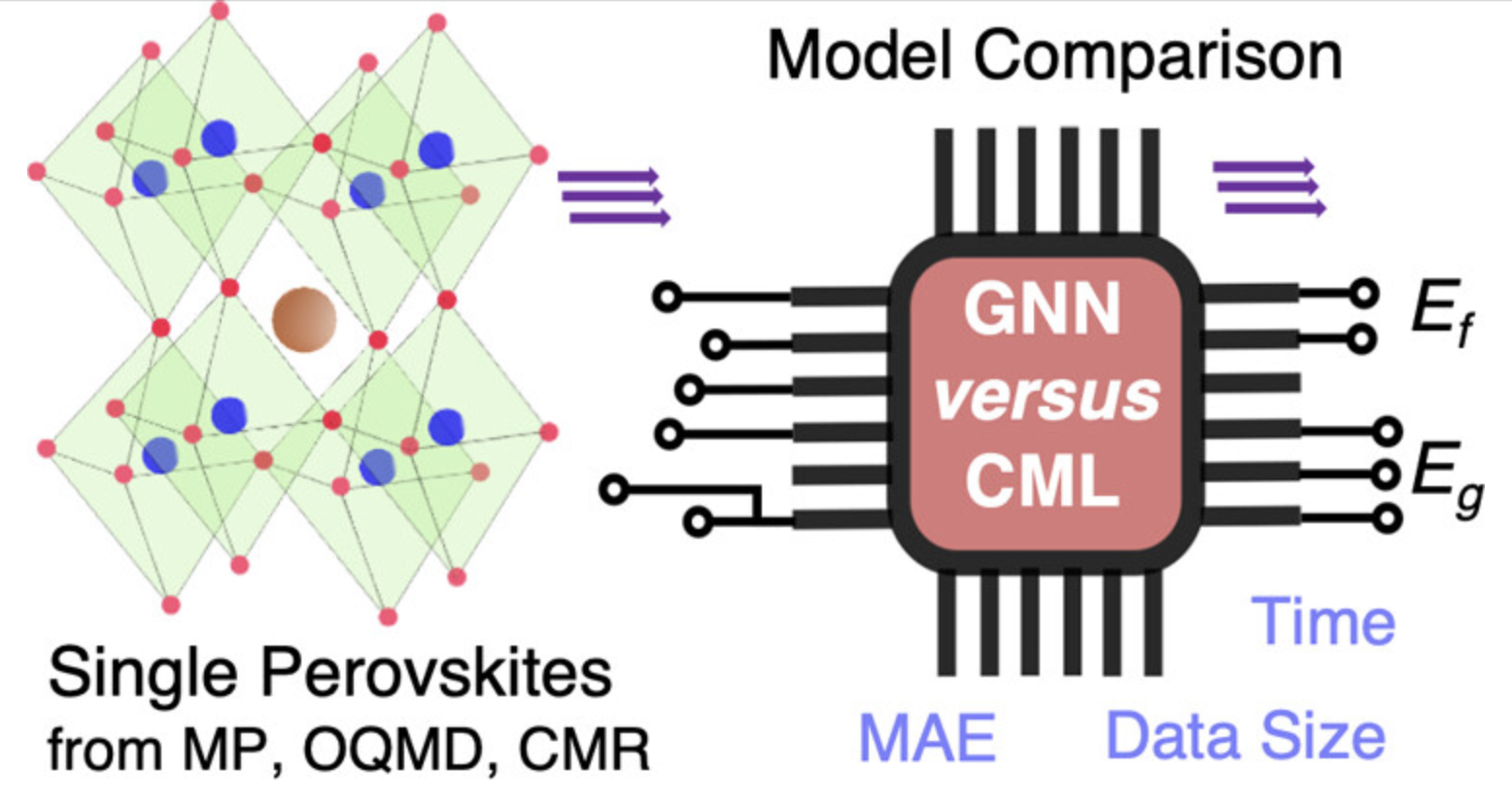
钙钛矿结构预测
预测钙钛矿材料的稳定性和光电性能。
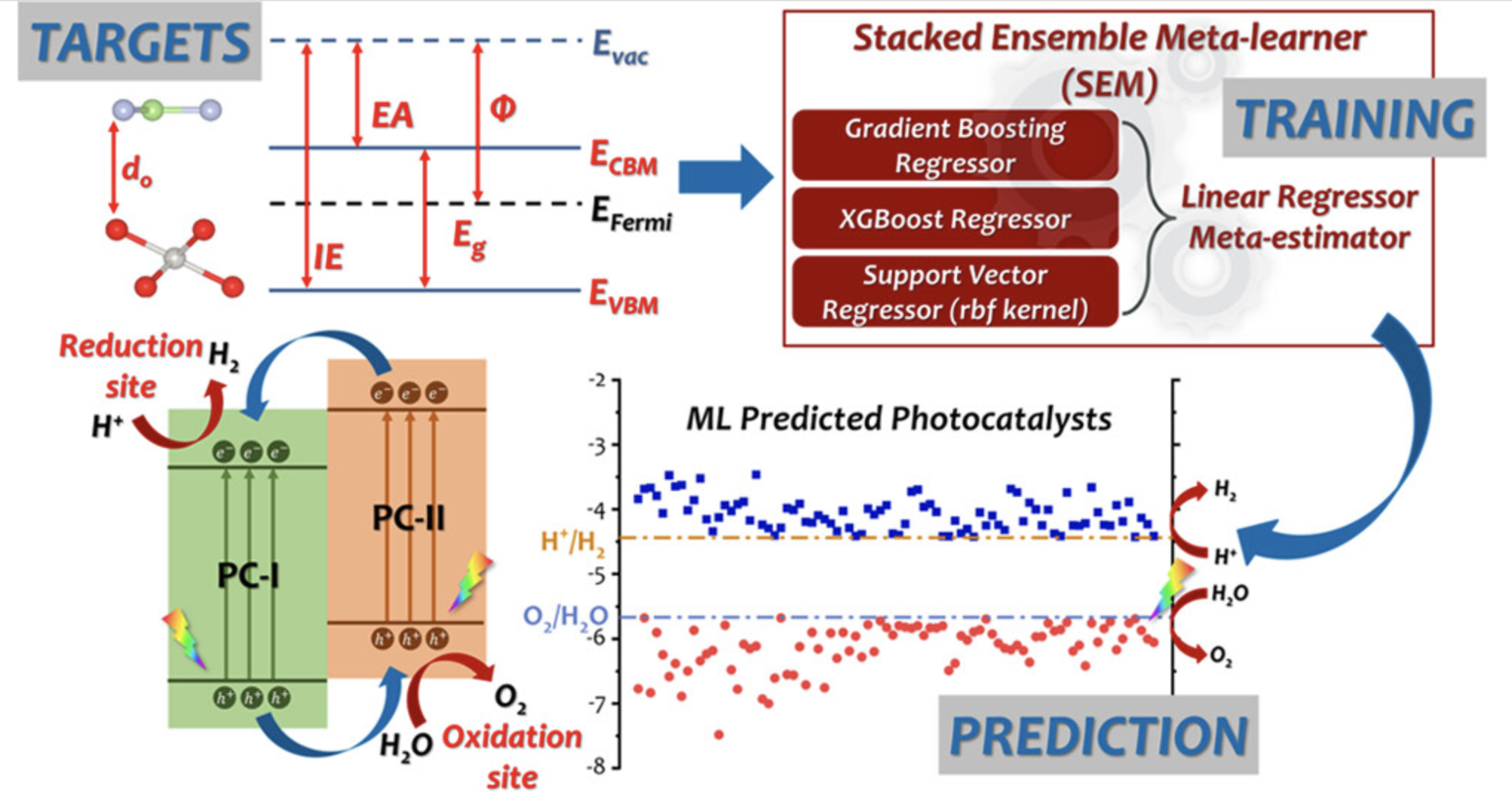
电极材料筛选
针对电池电极材料的成分优化和性能预测。
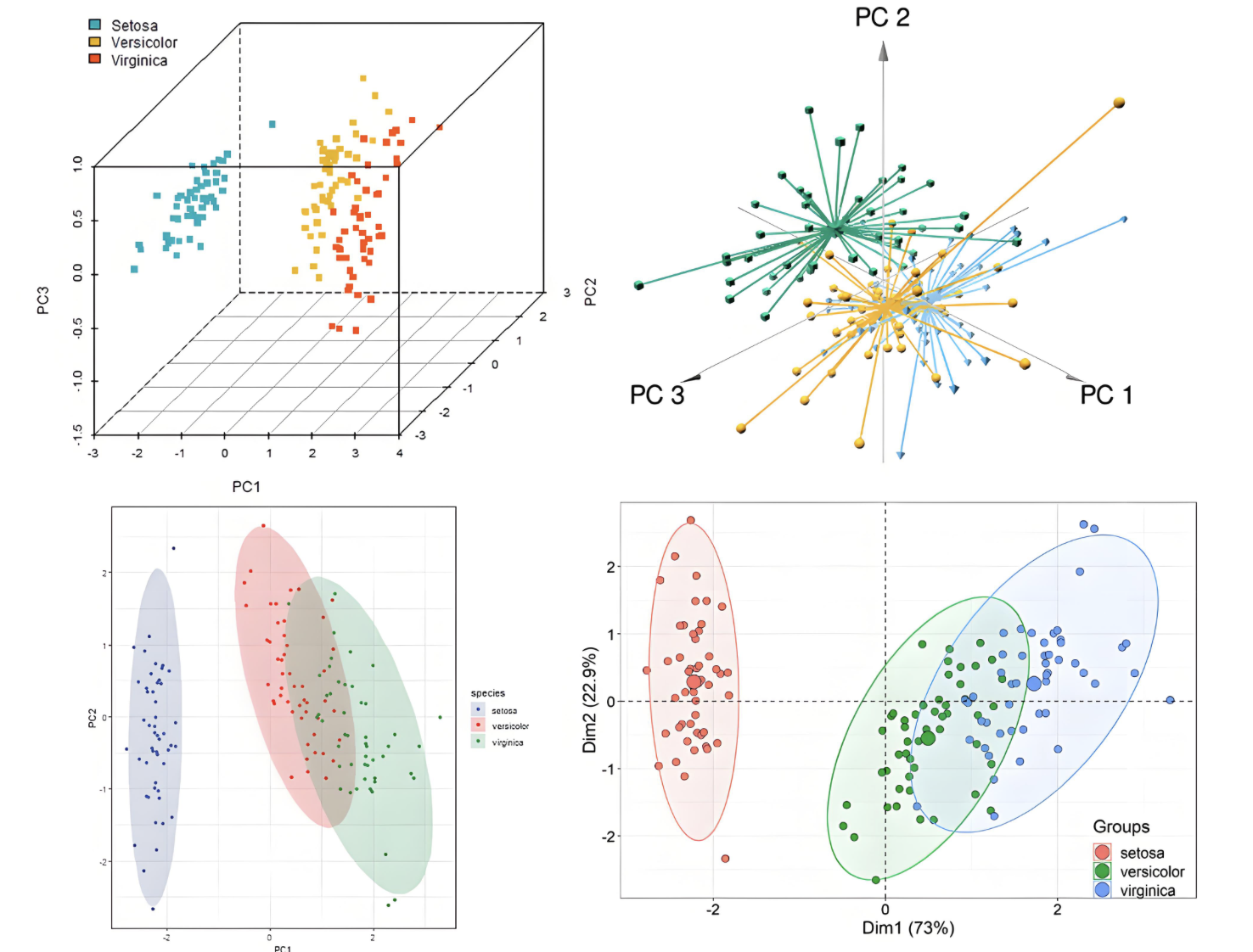
聚类分析
对材料数据进行聚类分析,发现相似结构和性质规律。
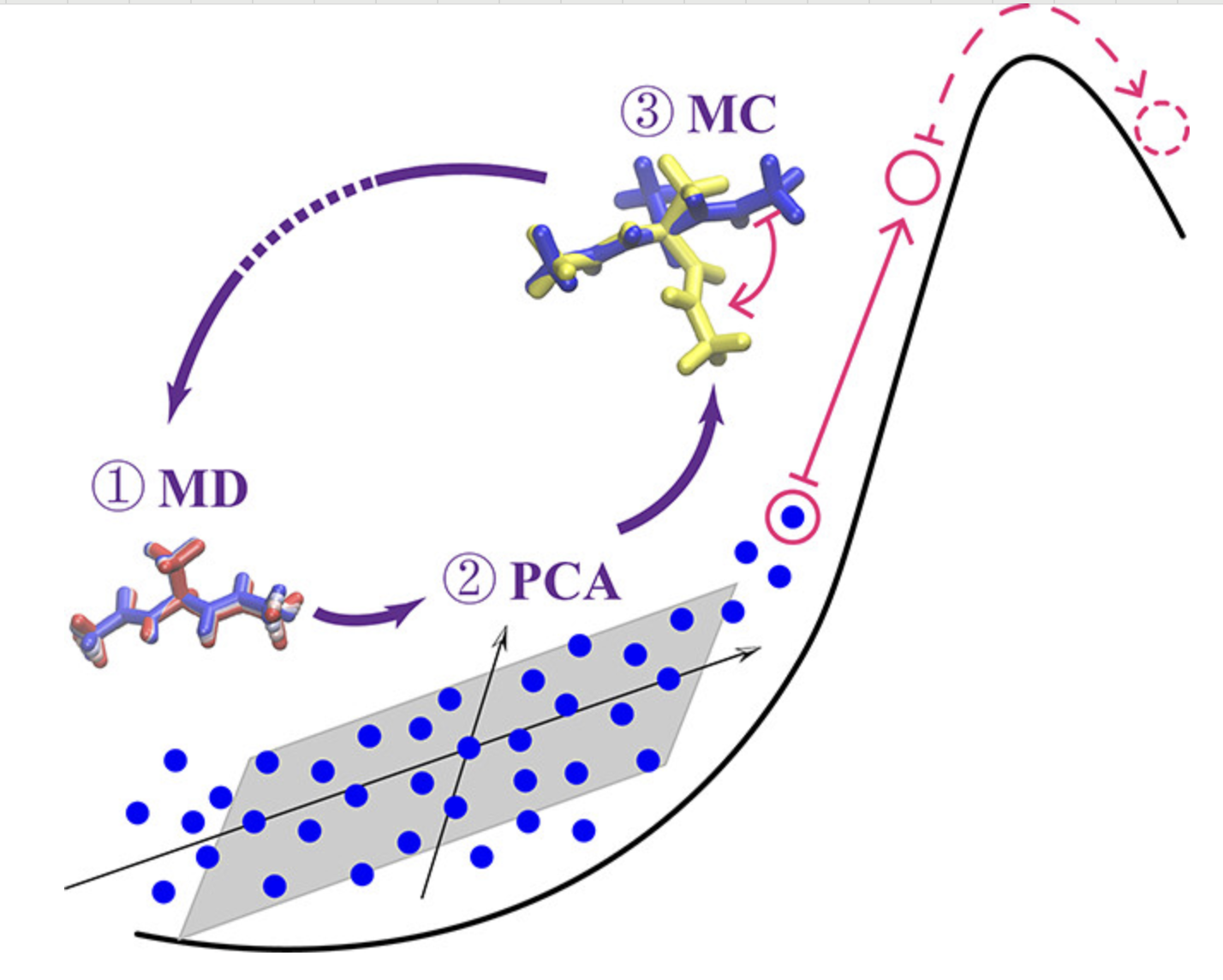
数据降维
使用PCA、t-SNE等方法对高维数据进行降维可视化。
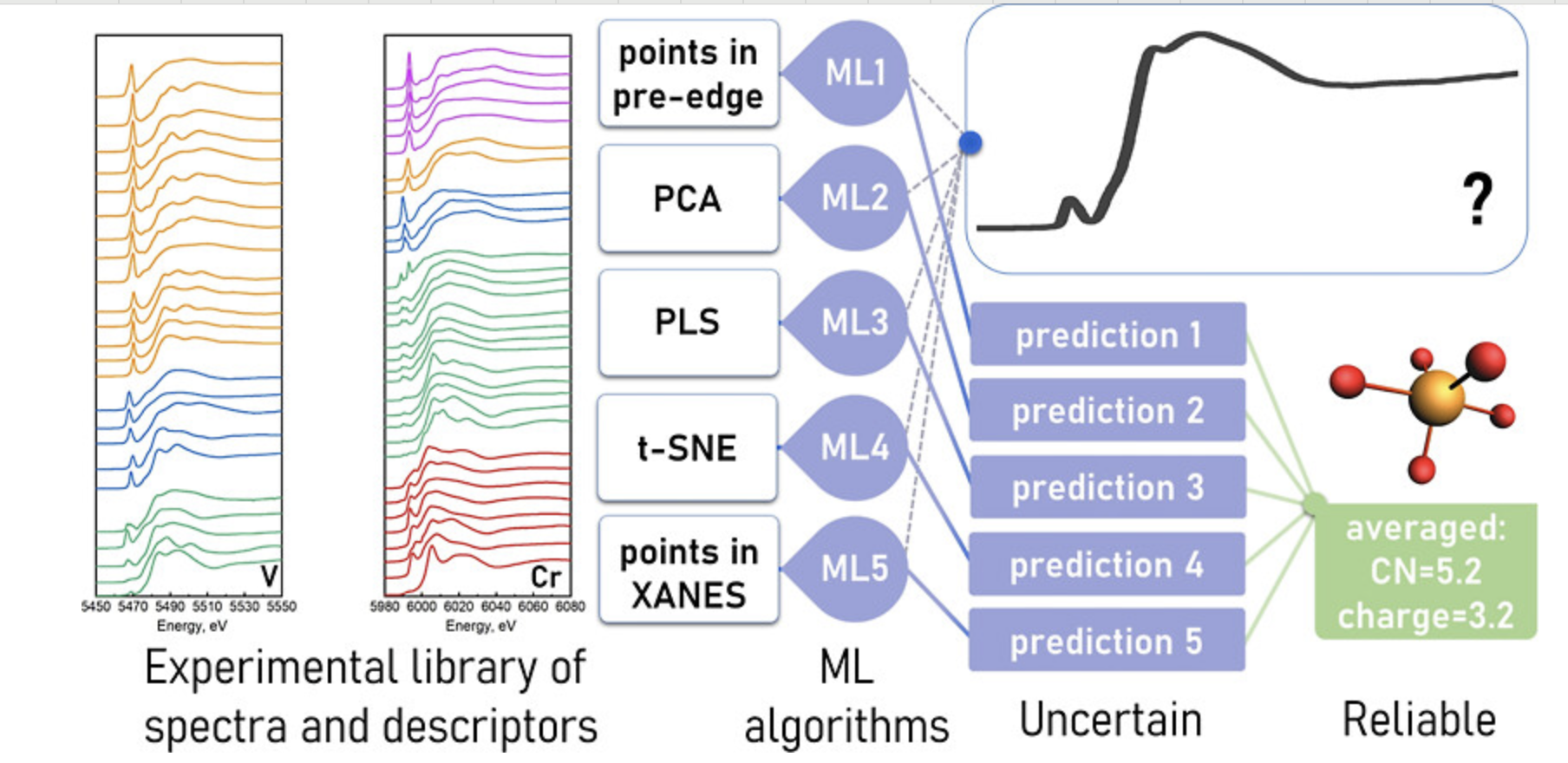
特征提取
从材料结构中提取有效的描述符和特征向量。
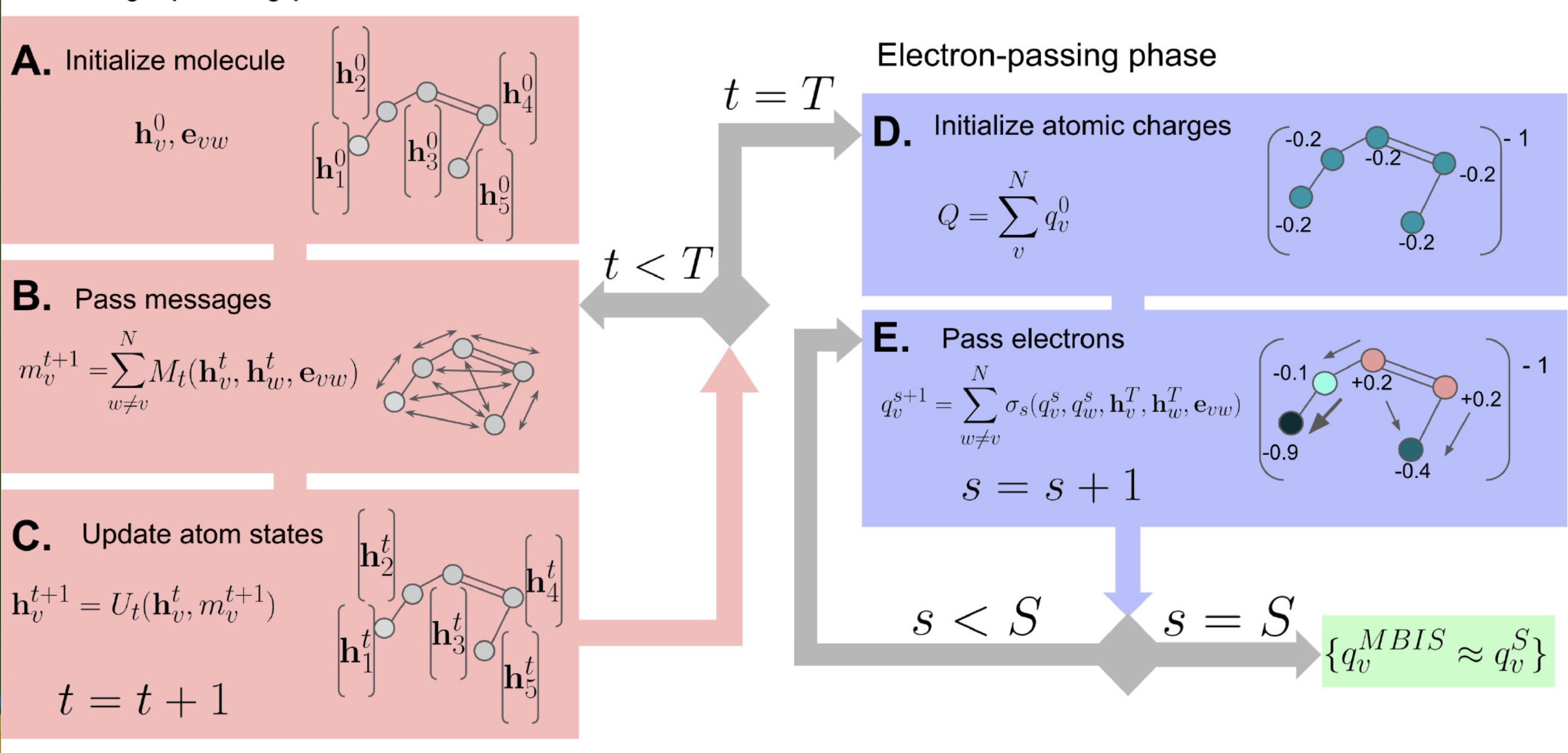
预测分子电荷
使用机器学习模型预测分子的原子电荷分布。
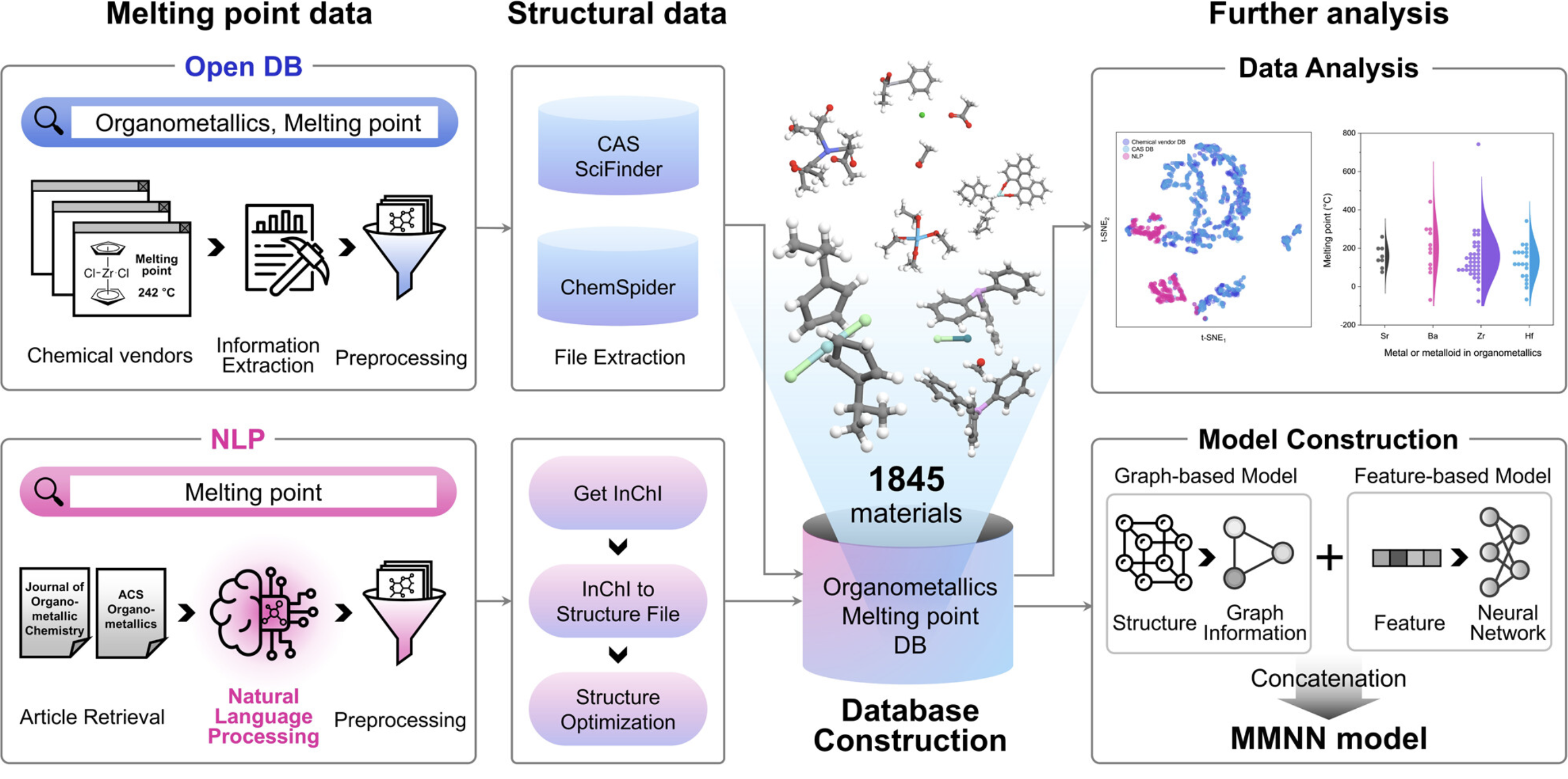
预测结构熔点
基于结构信息预测材料的熔点温度。
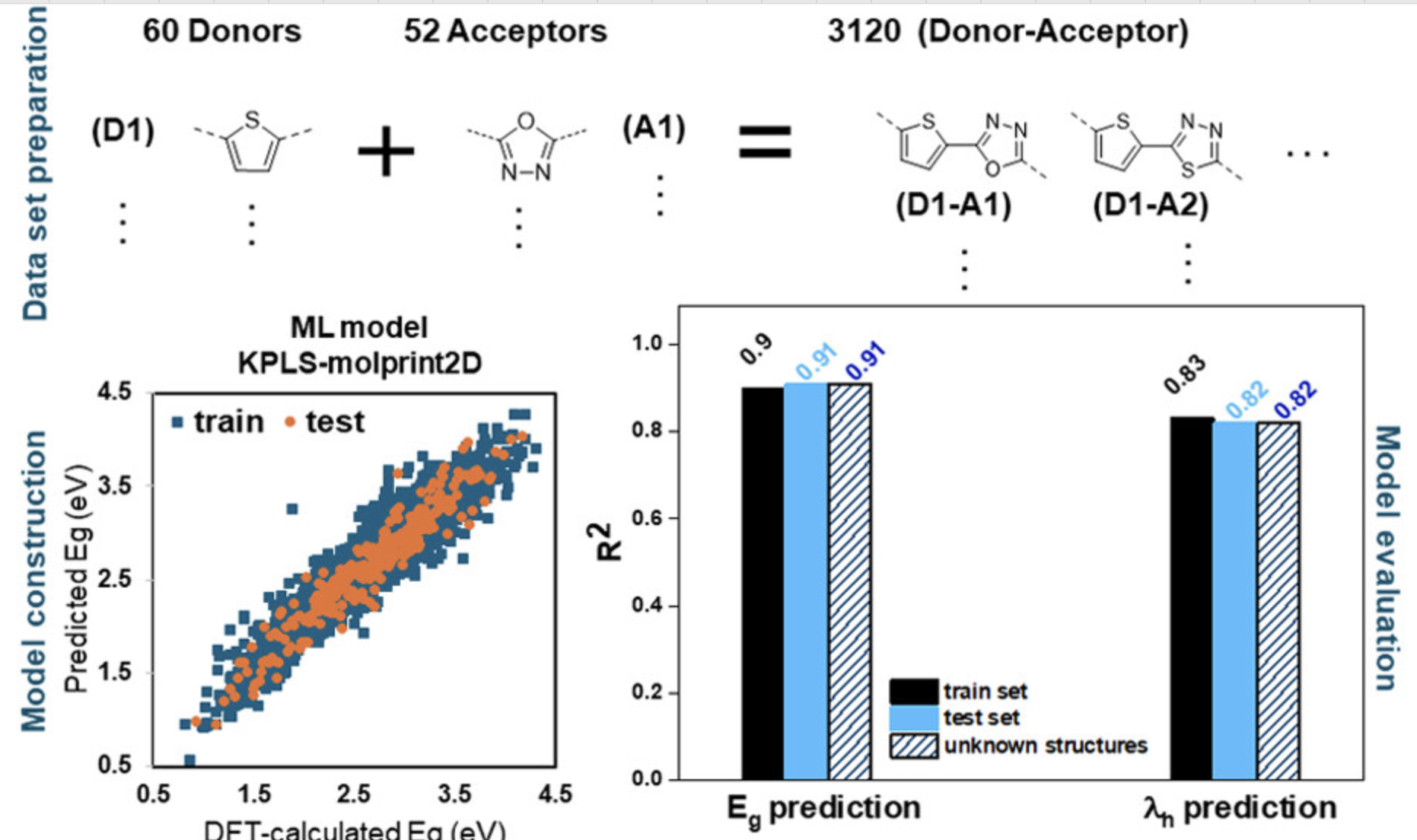
预测结构带隙
使用机器学习快速预测半导体材料的带隙。
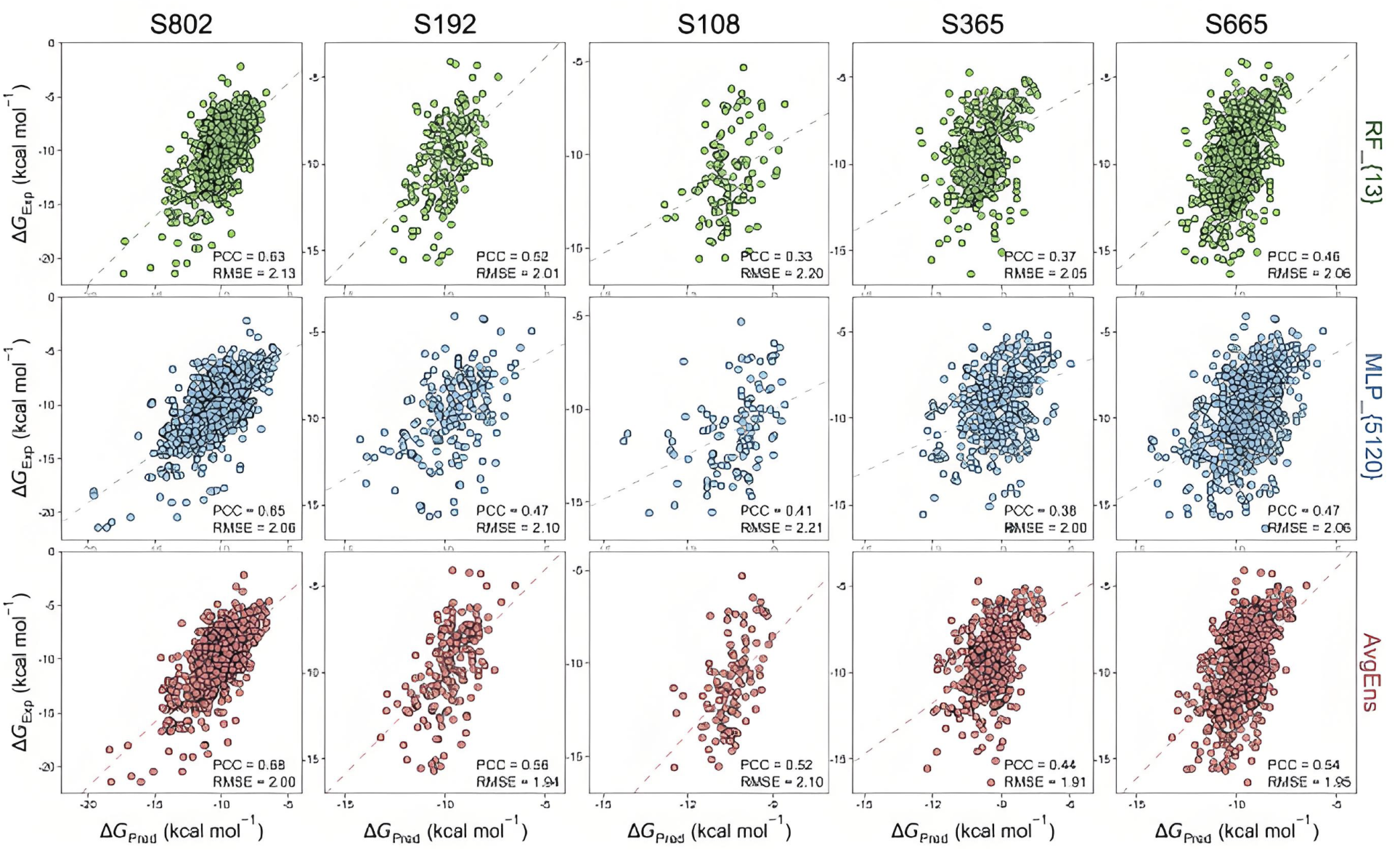
预测蛋白活性
基于蛋白质序列和结构预测其生物活性。
光谱分析
使用机器学习方法分析和预测各种光谱数据。
数据可视化
创建直观的图表和可视化界面展示计算结果。
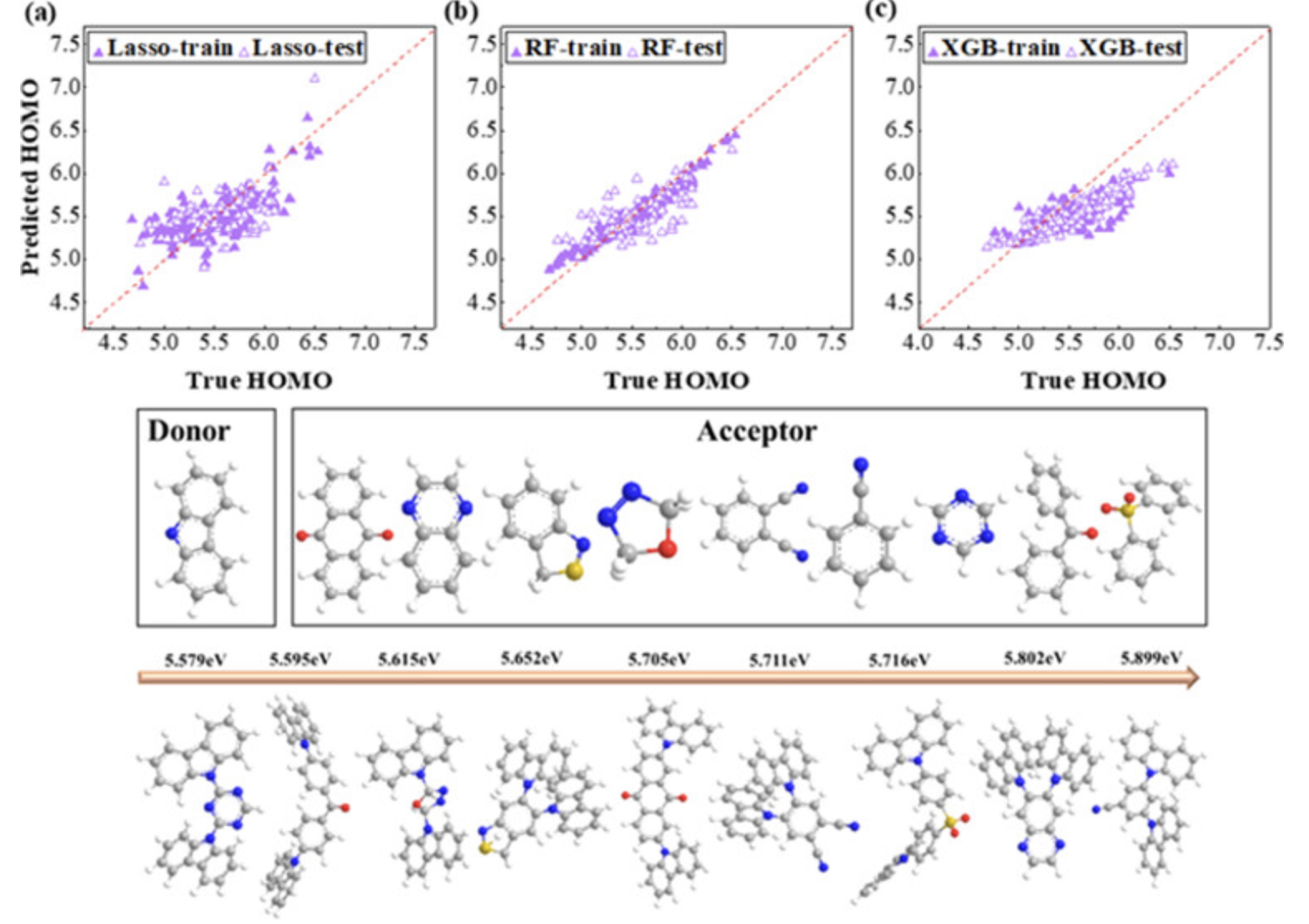
预测HOMO能级
使用机器学习预测分子的最高占据分子轨道能级。
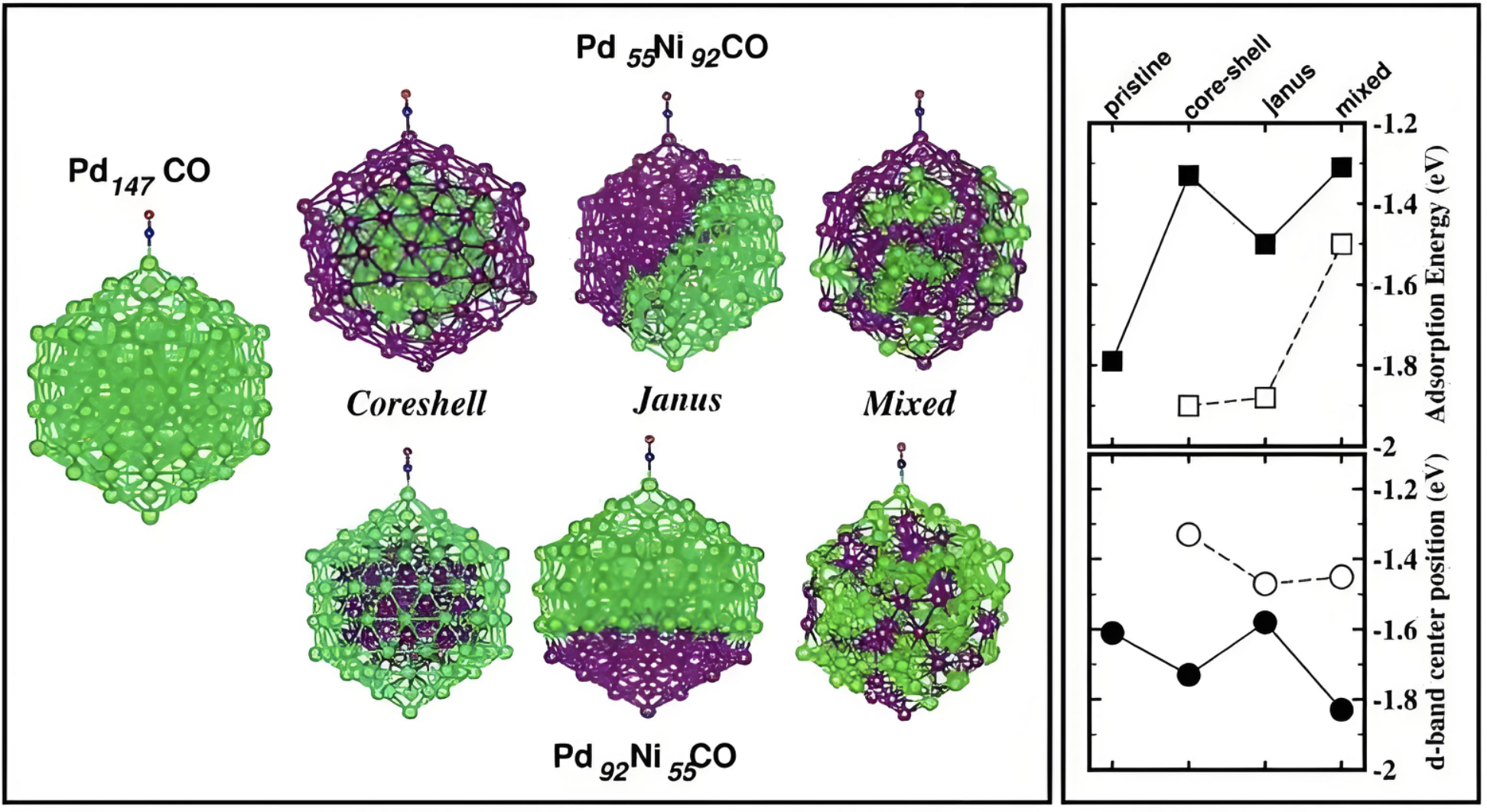
预测d带中心
快速预测过渡金属表面的d带中心位置。